Chemistry Editor, CBSE Evaluator | Updated on - Jun 23, 2026
The NCERT Solutions for Haloalkanes and Haloarenes will help you understand the steps involved in solving reaction-mechanism-based questions. Chapter 6 includes questions and answers on SN1, SN2, E1 and E2 mechanisms.
Haloalkanes and Haloarenes form the foundation of Organic Chemistry in Class 12. This page contains the 2026-27 NCERT Solutions PDF along with the exercise-wise breakdown, mechanism templates, and the latest PYQ map. The Solutions PDF provided in this article is aligned with the latest NCERT guidelines and CBSE-style answers.
You can find the complete NCERT Solutions for Haloalkanes and Haloarenes, including every in-text question, exercise problem, and CBSE-style reasoning answer, in the article below.
These NCERT Solutions are curated by subject experts, mapped to the 2026-27 NCERT, and refined against the last five years of CBSE Board, JEE Main, and NEET papers.
Haloalkanes and Haloarenes Exercise-by-Exercise Breakdown (NCERT Class 12 Chemistry)
The chapter packs 32 questions across in-text and back-exercise sets, skewed heavily toward mechanism comparison, conversion sequences, and reactivity-order reasoning. The table below maps each cluster to the sub-topic it tests.
The last twelve back-exercise questions are where most CBSE 3-mark and 5-mark answers originate, especially the conversion chains and SN1/SN2 reasoning sets.
What's Inside the Haloalkanes and Haloarenes NCERT Solutions PDF
The PDF contains every in-text and exercise problem from NCERT Chemistry Part II Chapter 6, fully solved with marker-ready answer structure.
32 questions solved: 10 in-text plus 22 back-exercise.
Mechanism diagrams for SN1, SN2, E1, E2 drawn step by step with transition states.
IUPAC nomenclature shown for every haloalkane and haloarene with branched-chain examples.
Reactivity-order reasoning blocks for vinyl, aryl, and alkyl halides with electron-density logic.
Distinction tests tabulated for primary, secondary, and tertiary halides.
Concept: The C-X bond is polar because halogens are more electronegative than carbon; this polarity is the reason haloalkanes undergo nucleophilic substitution, while haloarenes resist it due to partial double-bond character.
How will Collegedunia's NCERT Solutions Help You with Haloalkanes and Haloarenes?
Roughly 70 percent of CBSE questions from this chapter test the same four mechanism templates: SN1, SN2, E1, and E2. The solutions are written so you internalise the template structure while practising.
2026-27 NCERT Alignment: Every solution matches the current syllabus; the dropped polyhalogen-compound applications section is flagged so you don't waste time on it.
Mechanism-First Structure: Each conversion answer opens with the mechanism name, then shows the curly-arrow flow on a fresh line.
Expert Verification: Every IUPAC name and product structure cross-checked against the official NCERT key.
Common-Mistake Inline Notes: Saytzeff vs Hofmann elimination errors and chirality-inversion traps flagged inside each numerical.
Sample Fully-Solved Question: Class 12 Chemistry Chapter 6 Walk-Through
The walk-through below shows the answer shape a CBSE marker expects for a typical 3-mark SN1 vs SN2 distinction question. Copy this structure for any mechanism-comparison problem.
Question (3 marks). Predict the major product and mechanism when (CH3)3C-Br reacts with aqueous NaOH at room temperature, and justify your choice.
Step 1 (1M) - Identify substrate. The substrate is a tertiary alkyl halide. Tertiary carbocations are stabilised by hyperconjugation and inductive effects, so the SN1 pathway is favoured.
Step 2 (1M) - Mechanism. The C-Br bond ionises first to give (CH3)3C+ and Br-; the carbocation is then attacked by OH- from either face.
Step 3 (1M) - Product. The major product is tert-butyl alcohol, (CH3)3C-OH, with racemisation if the substrate had been chiral. Polar protic solvent (water) further stabilises the carbocation and accelerates SN1.
Watch Out: Writing only the product without naming the mechanism costs one mark; writing SN2 for a tertiary substrate costs two. Both slips drop a 3-mark answer to zero in many marking schemes.
Haloalkanes and Haloarenes Previous Year Questions Weightage (2021-2026)
The table tracks which sub-topic of Haloalkanes and Haloarenes was tested in CBSE Board, JEE Main, and NEET from 2021 onward. SN1/SN2 distinction and conversion sequences recur in almost every shift.
Year
CBSE Board
JEE Main
NEET
2026
Pending
SN1 vs SN2 reactivity order
Pending (exam rescheduled)
2025
Conversion: chlorobenzene to aniline (3M), Saytzeff product (2M)
Optical activity of 2-bromobutane
Reactivity of vinyl halide
2024
SN1 vs SN2 mechanism (5M)
Wurtz-Fittig reaction product
Haloarene resonance stabilisation
2023
Finkelstein reaction (2M), IUPAC of branched haloalkane (2M)
Reactivity order of alkyl halides
Markovnikov vs anti-Markovnikov
2022
Conversion sequence (3M), E1 vs E2 (3M)
SN1 carbocation stability
DDT and freon uses
2021
Polarity of C-X bond (2M)
-
Aryl halide vs alkyl halide reactivity
SN1 vs SN2 reasoning appeared in 4 of the last 5 CBSE Board papers - the single most-repeated 3-marker of this chapter.
How to Study Haloalkanes and Haloarenes for Class 12 Chemistry Boards
Plan roughly 9 to 11 hours of focused practice across four sessions.
Session 1 (2h) - Nomenclature and classification. NCERT 6.1 to 6.2; in-text Q6.1 to Q6.4 and back exercise Q6.1 to Q6.5.
Session 2 (3h) - Preparation and physical properties. Sandmeyer, Finkelstein, Hunsdiecker, free-radical halogenation; solve Q6.6 to Q6.12.
Session 3 (3h) - SN1, SN2, E1, E2 mechanisms. Build a one-page comparison sheet; solve Q6.13 to Q6.18.
Session 4 (3h) - Haloarenes and conversions. Electrophilic substitution in haloarenes, multi-step conversions; solve Q6.19 to Q6.22.
Quick Tip: With only 4 hours left before the exam, drill the SN1 vs SN2 comparison table and three conversion chains (chlorobenzene to aniline, alkyl halide to alkene, haloalkane to nitrile). They cover the bulk of the 3-mark and 5-mark questions every year.
Haloalkanes and Haloarenes Top 5 Reactions for Quick Recall
The five reactions below recur in CBSE Board, JEE Main, and NEET questions on this chapter. The full master sheet with named-reaction templates sits on the Collegedunia Formula Sheet.
Common Mistakes Students Make in Haloalkanes and Haloarenes
Five answer-writing slips turn a full-marks answer into a half-marks one in CBSE scripts.
Predicting SN2 for tertiary substrates.Tertiary always favours SN1 in polar protic solvent; getting this wrong costs both marks.
Skipping the curly-arrow flow. Mechanism marks need explicit electron-pair movement, not just the product name.
Forgetting Saytzeff vs Hofmann.Saytzeff for E1/E2 with small bases; Hofmann for bulky bases. Mixing them is a 2-mark slip.
Naming aryl halide reactivity wrong. Aryl halides are less reactive due to partial double-bond character; writing "more reactive" is an instant zero.
Wrong IUPAC numbering. The lowest locant goes to the halogen, not to the longest chain end. A wrong locant set drops 1 of 2 marks.
Haloalkanes and Haloarenes Topic-by-Topic Summary for 12th Chemistry
The nine sub-topics below form the spine of the chapter; the full deep-walk with worked illustrations sits on the Collegedunia Notes page.
Classification and nomenclature: Mono-, di-, polyhalogen compounds; primary, secondary, tertiary; IUPAC naming with halogen as substituent.
Methods of preparation: From alcohols (with HX, PX3, SOCl2, and conc. HCl with anhydrous ZnCl2 in the Lucas test), alkenes (Markovnikov HX, anti-Markovnikov HBr via the Kharasch peroxide effect), and hydrocarbons; halogen-exchange via Finkelstein and Swarts reactions.
Bond length and C-X polarity: Bond length C-F < C-Cl < C-Br < C-I; dipole moment of haloalkanes peaks at C-Cl, not C-F, because charge separation grows with bond length.
Markovnikov vs anti-Markovnikov addition: Markovnikov gives the more substituted halide; the Kharasch (peroxide) effect reverses regioselectivity for HBr only.
Chemical reactions of haloalkanes: Nucleophilic substitution (SN1, SN2), elimination (E1, E2 with Saytzeff vs Hofmann competition), reaction with metals (Wurtz, Grignard).
Chemical reactions of haloarenes: Chlorobenzene reactivity is limited (resonance shortens and strengthens the Ar-X bond); electrophilic substitution with directive effect, reaction with metals (Fittig, Wurtz-Fittig).
Optical isomerism, chiral centre identification and R/S configuration: Chiral haloalkanes such as 2-bromobutane are assigned using CIP rules; SN1 racemisation versus SN2 inversion is a recurring CBSE question.
Polyhalogen compounds: Uses of chloroform, iodoform, CCl4, DDT (preparation from chlorobenzene + chloral), freons / CFCs (Swarts on CCl4); bulk industrial applications dropped in the new edition.
Haloalkanes and Haloarenes Weightage Compared Across Class 12 Chemistry Chapters
The visual maps the typical CBSE marks distribution across all 10 chapters of NCERT Chemistry, averaged over the last five board papers. Haloalkanes and Haloarenes sits in the mid band, with two strong PYQ-recurrence streaks per year.
Ch 1 Solutions
6 marks
Ch 2 Electrochemistry
7 marks
Ch 3 Chemical Kinetics
7 marks
Ch 4 d- and f-Block Elements
7 marks
Ch 5 Coordination Compounds
8 marks
Ch 6 Haloalkanes and Haloarenes
6 marks
Ch 7 Alcohols, Phenols and Ethers
7 marks
Ch 8 Aldehydes, Ketones and Acids
8 marks
Ch 9 Amines
6 marks
Ch 10 Biomolecules
5 marks
All NCERT Solutions for Haloalkanes and Haloarenes with Step-by-Step Working
Every NCERT textbook question for Class 12 Chemistry Chapter 6 Haloalkanes and Haloarenes is listed below with its full Solution and Expert Solution hidden inside collapsible tabs. Click Check Solution to reveal the step-by-step working; click Expert Solution for the expanded explanation.
Questions
Q 6.1
Name the following halides according to IUPAC system and classify them as alkyl, allyl, benzyl (primary, secondary, tertiary), vinyl or aryl halides:
(i) (CH3)2CHCH(Cl)CH3
(ii) CH3CH2CH(CH3)CH(C2H5)Cl
(iii) CH3CH2C(CH3)2CH2I
(iv) (CH3)3CCH2CH(Br)C6H5
(v) CH3CH(CH3)CH(Br)CH3
(vi) CH3C(C2H5)2CH2Br
(vii) CH3C(Cl)(C2H5)CH2CH3
(viii) CH3CH=C(Cl)CH2CH(CH3)2
(ix) CH3CH=CHC(Br)(CH3)2
(x) p-ClC6H4CH2CH(CH3)2
(xi) m-ClCH2C6H4CH2C(CH3)3
(xii) o-Br-C6H4CH(CH3)CH2CH3
Concept used. The IUPAC name of a haloalkane is built from four pieces, in order:
[label=(),leftmargin=*,itemsep=1pt]
Parent chain: the longest continuous carbon chain that contains the carbon carrying the halogen.
Numbering: number the parent chain from the end that gives the lowest locant to the first point of difference (halogen vs. alkyl substituents are treated equally; the lower set wins).
Substituent prefixes: name halogens as fluoro-, chloro-, bromo-, iodo-, alkyls as methyl, ethyl, , alphabetically (di-, tri- do not count for alphabetisation).
Suffix: -ane for the parent alkane, -ene/-yne for unsaturated chains, and the locants of double/triple bonds get the lowest set after substituent locants are tied.
Classification rules.
[leftmargin=*,itemsep=1pt]
Alkyl halide: X on an sp3 carbon of an open chain. It is primary (1∘) if the carbon carrying X is bonded to one other carbon, secondary (2∘) if bonded to two, tertiary (3∘) if bonded to three.
Allyl halide: X on an sp3 carbon next to C=C (i.e. on the allylic carbon).
Benzyl halide: X on the sp3 carbon directly attached to a benzene ring.
Vinyl halide: X on an sp2 carbon of C=C.
Aryl halide: X attached directly to an aromatic sp2 ring carbon.
The ``lowest locant'' rule
If both numbering directions give the same first locant, compare the second, then third, etc. If all locants are equal, the alphabetically earlier substituent gets the smaller number.
(i) (CH3)2CHCH(Cl)CH3. Expand: CH3-CH(CH3)-CH(Cl)-CH3. Longest chain through the C–Cl carbon = 4 carbons (butane). Numbering from the right gives Cl at C2 and CH3 substituent at C3; chloro alphabetises before methyl, so chloro gets the smaller locant. Name: 2-chloro-3-methylbutane. The Cl-bearing carbon is attached to two other carbons, hence secondary (2∘) alkyl halide.
(ii) CH3CH2CH(CH3)CH(C2H5)Cl. The longest chain through C-Cl runs through the ethyl branch: CH3-CH2-CH(CH3)-CH(Cl)-CH2-CH3 (6 C, hexane). Numbering from the Cl end gives Cl at C3 and CH3 at C4. Name: 3-chloro-4-methylhexane. The Cl-bearing carbon is bonded to two other carbons, hence secondary (2∘) alkyl halide.
(iii) CH3CH2C(CH3)2CH2I. Longest chain = I-CH2-C(CH3)2-CH2-CH3 (4 C through I; the second CH3 on the quaternary carbon becomes a methyl substituent). Numbering from the I end gives I at C1 and two methyls at C2. Name: 1-iodo-2,2-dimethylbutane. The I-carbon is bonded to one other carbon, hence primary (1∘) alkyl halide.
(iv) (CH3)3CCH2CH(Br)C6H5. The Br sits on an sp3 carbon directly attached to a benzene ring. Choose the benzene ring as the parent. Substituent: -CH(Br)CH2C(CH3)3 which is 1-bromo-3,3-dimethylbutyl. Name: (1-bromo-3,3-dimethylbutyl)benzene. Because Br is on the carbon directly attached to Ph, this is a secondary (2∘) benzyl halide.
(v) CH3CH(CH3)CH(Br)CH3. Longest chain through C-Br = 4 C (butane). Numbering from the right gives Br at C2 and CH3 at C3. Bromo alphabetises before methyl, so bromo gets the smaller locant. Name: 2-bromo-3-methylbutane. The Br-carbon is bonded to two other carbons, hence secondary (2∘) alkyl halide.
(vi) CH3C(C2H5)2CH2Br. Choose the longest chain through C-Br: Br-CH2-C(CH3)(C2H5)-C2H5 (4 C, butane). The remaining C2H5 and CH3 become substituents at C2. Name: 1-bromo-2-ethyl-2-methylbutane. Br-carbon bonded to one other carbon, hence primary (1∘) alkyl halide.
(vii) CH3C(Cl)(C2H5)CH2CH3. Longest chain through C-Cl = 5 C (pentane). Numbering puts Cl at C3 with a CH3 also at C3. Name: 3-chloro-3-methylpentane. The Cl-carbon is bonded to three other carbons, hence tertiary (3∘) alkyl halide.
(viii) CH3CH=C(Cl)CH2CH(CH3)2. Parent chain = 6 C with one C=C. Numbering from the left puts C=C between C2 and C3, Cl at C3, CH3 at C5. Name: 3-chloro-5-methylhex-2-ene. The Cl sits on an sp2 carbon of C=C, hence vinyl halide.
(ix) CH3CH=CHC(Br)(CH3)2. Parent chain = 5 C with one C=C. Numbering must give the C=C (the principal characteristic) the lowest locant, so number from the left: CH3(C1)-CH(C2)=CH(C3)-C(Br)(CH3)(C4)-CH3(C5). C=C between C2 and C3 (locant 2 beats 3), Br at C4 and a methyl substituent at C4. Name: 4-bromo-4-methylpent-2-ene. The Br-carbon is sp3 and adjacent to C=C, hence allyl halide (and a tertiary one, since it is bonded to three other carbons).
(x) p-ClC6H4CH2CH(CH3)2. Parent: benzene ring; the substituent -CH2-CH(CH3)2 is 2-methylpropyl (isobutyl). Name: 1-chloro-4-(2-methylpropyl)benzene. The Cl is on an aromatic ring carbon, hence aryl halide.
(xi) m-ClCH2C6H4CH2C(CH3)3. The Cl sits on a benzylic -CH2- group. Substituents on the ring: -CH2Cl (chloromethyl) and -CH2C(CH3)3 (2,2-dimethylpropyl). meta relationship = 1,3. Name: 1-(chloromethyl)-3-(2,2-dimethylpropyl)benzene. Cl on a carbon directly bonded to the ring, hence primary (1∘) benzyl halide.
(xii) o-Br-C6H4CH(CH3)CH2CH3. The side chain is butan-2-yl (sec-butyl) and the ring carries Br ortho (1,2) to it. Name: 1-bromo-2-(butan-2-yl)benzene. Br on the aromatic ring, hence aryl halide.
See the bold IUPAC name and class type stated in each step above.
AM
Aarav Mehta
M.Sc Chemistry, IIT Kanpur
Verified Expert
Structural observation. Treat naming as a two-pass problem: first pick the parent (longest chain through the halogen for aliphatics, the ring for aromatics with short branches), then assign locants. Classification is independent of the IUPAC name: just look at the hybridisation of the C attached to X and what else is on it.
[leftmargin=*,itemsep=1pt]
Alkyl/Benzyl/Allyl: X is on an sp3 carbon.
Vinyl/Aryl: X is on an sp2 carbon.
Further split sp3 into 1∘/2∘/3∘ by counting carbons on the C–X carbon.
Alternative approach: classification first. A useful shortcut for board questions is to do classification before naming. Drawing the carbon skeleton already tells you the hybridisation of the C–X carbon, and one glance gives 1∘/2∘/3∘, allyl, benzyl, vinyl or aryl. Once classified, the IUPAC name becomes almost mechanical: aryl/vinyl need the unsaturation/ring in the parent, allyl/benzyl give either choice of parent, and alkyl is always named as an alkane.
Group the questions: (i), (ii), (v), (vii) are open-chain sp3 halides, so they are named as substituted alkanes. (iii), (vi) have a quaternary carbon adjacent to CH2X; the longest chain runs through one of the long arms, the other becomes a substituent.
(iv), (xi) have the halogen on the benzylic CH2 or CHR: benzyl class. Their names use benzene as the parent because the side chain is short.
(viii) has Cl on sp2 C=C carbon, so vinyl. (ix) has Br on an sp3 carbon directly attached to C=C, so allyl. Watch the distinction: it is not the geometry of the bond, it is which carbon (allylic = next-door to C=C; vinylic = part of C=C).
(x), (xii) have X on the ring carbon itself, so aryl. The chains hanging off the ring are substituents, named as isobutyl (= 2-methylpropyl) and sec-butyl (= butan-2-yl).
Re-read every assigned name to check alphabetical order of prefixes, lowest locants, correct -ene/-yne for unsaturated chains.
Reactivity preview (concept linkage). The class label tells you how the molecule will behave later in the chapter: 3∘ alkyl and benzyl/allyl tertiary go through SN1; 1∘ alkyl go through SN2; vinyl and aryl halides are nearly unreactive toward nucleophilic substitution because the C–X carbon is sp2 and the bond has partial double-bond character.
Why this matters. The two questions ``what is the IUPAC name?'' and ``what type of halide is it?'' are independent and need two passes over the structure. JEE/NEET routinely test the allyl vs. vinyl and benzyl vs. aryl distinction because one carbon shift changes everything (reactivity, stability, mechanism). A typical 2-mark CBSE question asks ``classify the following halide and write its IUPAC name'' — both parts get full marks only if the class label matches what you draw.
Give the IUPAC names of the following compounds:
(i) CH3CH(Cl)CH(Br)CH3
(ii) CHF2CBrClF
(iii) ClCH2C#CCH2Br
(iv) (CCl3)3CCl
(v) CH3C(p-ClC6H4)2CH(Br)CH3
(vi) (CH3)3CCH=CClC6H4I-p
Concept used. For polyhalogenated compounds, treat each halogen as a separate substituent and list them alphabetically (bromo, chloro, fluoro, iodo). For chains with multiple halogens, give the lowest set of locants to the substituents collectively. When the parent has a triple bond, use the suffix -yne.
(i) CH3CH(Cl)CH(Br)CH3. Parent: butane (4 C). Numbering from either end gives substituents at C2 and C3, a tie. Bromo alphabetises before chloro, so Br must get the smaller locant. Number from the right. Name: 2-bromo-3-chlorobutane.
(ii) CHF2CBrClF. Parent: ethane (2 C). Number from the right so the larger cluster of substituents gets the lower locant. C1 carries Br, Cl, F; C2 carries F, F. Name: 1-bromo-1-chloro-1,2,2-trifluoroethane.
(iii) ClCH2C#CCH2Br. Parent: 4 C with a triple bond, i.e. but-2-yne. Bromo alphabetises before chloro, so Br gets locant 1. Name: 1-bromo-4-chlorobut-2-yne.
(iv) (CCl3)3CCl. The central carbon is bonded to three CCl3 groups and one Cl. The longest chain through it is 2 C (ethane). Substituents: three Cl on C2 (2,2,2-trichloro), one Cl on C1 and two CCl3 (trichloromethyl) on C1. Alphabetise on the substituent root: chloro (c) before trichloromethyl (t). Name: 1,2,2,2-tetrachloro-1,1-bis(trichloromethyl)ethane.
(v) CH3C(p-ClC6H4)2CH(Br)CH3. Parent: butane. From the right, Br at C2; two p-chlorophenyl groups at C3. Name: 2-bromo-3,3-bis(4-chlorophenyl)butane.
(vi) (CH3)3CCH=CClC6H4I-p. The longest chain through C=C runs through the tert-butyl carbon out to the =CCl: 4 C (but-1-ene). The p-iodophenyl sits on C1, Cl on C1, and the tert-butyl's three methyls put two more methyls and the chain end on C3. Name: 1-chloro-1-(4-iodophenyl)-3,3-dimethylbut-1-ene.
Strategic angle. Polyhalogen names are a discipline problem, not a chemistry problem. Step through three checks: (a) identify the parent chain, (b) number so the substituents take the lowest set, (c) write substituents alphabetically.
Alphabetisation rule, expanded. When two substituents tie for the locant, the first letter of the substituent root (not the multiplying prefix) decides which gets the smaller number. So dimethyl alphabetises as ``m''; trichloromethyl alphabetises as ``t''; bis(4-chlorophenyl) alphabetises as ``c'' (chlorophenyl). For (iv), the substituent trichloromethyl comes after chloro alphabetically, so chloro locants are written first.
For (i), the chain is short and the two halogens differ: bromo alphabetises before chloro, so it must take the smaller locant. Number from the right.
For (ii), the parent is ethane. Pick the end with the larger substituent cluster. 1-Br + 1-Cl + 1-F + 2,2-(F,F) gives three F atoms, so the trifluoro prefix is 1,2,2.
For (iii), the locant of the triple bond and the locant of the lowest-set substituents are both 2: tie. Use alphabet: bromo < chloro, so bromo gets the 1 position.
For (iv), the chain is just two carbons. Three CCl3 groups become three trichloromethyl substituents. Locants: 1,2,2,2-tetrachloro plus 1,1-bis(trichloromethyl). Note ``bis'' (not ``di'') because the substituent name trichloromethyl is itself a compound name.
For (v) and (vi), the aryl groups go in parentheses with their substitution locant: 4-chlorophenyl, 4-iodophenyl. In (vi) the C=C is between C1 and C2 of a but-1-ene chain.
Cross-check. Re-read each name and rebuild the structure on paper; if you can recover the input formula unambiguously, the name is correct. This back-translation is the surest way to catch wrong locants.
Why this matters. The 2026–27 syllabus emphasises modern IUPAC formatting (locants attached to the suffix, e.g. ``but-2-ene'' not ``2-butene''), and numbering questions appear every year in board papers as 1- and 2-mark questions.
Same six names as in the main solution.
Q 6.3
Write the structures of the following organic halogen compounds:
(i) 2-Chloro-3-methylpentane
(ii) p-Bromochlorobenzene
(iii) 1-Chloro-4-ethylcyclohexane
(iv) 2-(2-Chlorophenyl)-1-iodooctane
(v) 2-Bromobutane
(vi) 4-tert-Butyl-3-iodoheptane
(vii) 1-Bromo-4-sec-butyl-2-methylbenzene
(viii) 1,4-Dibromobut-2-ene
Concept used. To turn a name into a structure we work backwards through the IUPAC rules: read the parent (chain length, saturated/unsaturated, cyclic), then place substituents at their locants, then check that the locants are the lowest possible set.
(i) 2-Chloro-3-methylpentane. Pentane = 5 C chain. Place Cl at C2 and CH3 at C3: CH3-CH(Cl)-CH(CH3)-CH2-CH3.
(ii) p-Bromochlorobenzene.para means 1,4 disubstitution on a benzene ring; place Br at C1 and Cl at C4: Br-C6H4-Cl (1,4).
(iii) 1-Chloro-4-ethylcyclohexane. Cyclohexane has 6 ring carbons; place Cl at C1 and C2H5 at C4 (across the ring).
(iv) 2-(2-Chlorophenyl)-1-iodooctane. Octane = 8 C chain; I at C1, and at C2 we attach 2-chlorophenyl (a benzene ring with Cl at its ortho position relative to the link): I-CH2-CH(o-ClC6H4)-CH2-CH2-CH2-CH2-CH2-CH3.
(v) 2-Bromobutane. Butane = 4 C; Br at C2: CH3-CH(Br)-CH2-CH3.
(vi) 4-tert-Butyl-3-iodoheptane. Heptane = 7 C; I at C3 and -C(CH3)3 at C4: CH3CH2-CH(I)-CH(C(CH3)3)-CH2-CH2-CH3.
(vii) 1-Bromo-4-sec-butyl-2-methylbenzene. Benzene ring; Br at C1, CH3 at C2, sec-butyl (= CH(CH3)CH2CH3) at C4.
(viii) 1,4-Dibromobut-2-ene. But-2-ene means a 4 C chain with C=C between C2 and C3; place Br at C1 and C4: BrCH2-CH=CH-CH2Br.
!%
[See diagram in the PDF version]
Structures drawn step-by-step above.
RB
Riya Banerjee
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Picture-first. Name → structure is the inverse of naming. Read the parent first (chain length and saturation), then walk through the locants and attach substituents at the named positions. Treat aryl substituents like 2-chlorophenyl as sub-named groups: a phenyl ring with a chloro at the ortho (2-) position.
Decoding nested substituent names. When a name has a substituent in parentheses, both the name outside and inside need parsing. ``2-(2-chlorophenyl)-1-iodooctane'' decodes as: parent = octane (8 C); at C1 attach iodine; at C2 attach ``2-chlorophenyl'' (a phenyl ring where its own C2 carries Cl). The parenthesised number always refers to the substituent's own numbering, not the parent chain's.
For (i), (v), (vi), (viii) the parent is an open-chain alkane/alkene. Draw the chain, number left to right by default, drop substituents at the listed locants.
For (ii), (vii) the parent is benzene. Reduce the substituent names to fragments (Br, Cl, CH3, sec-butyl). Place at the listed ring positions. Remember para= 1,4 and ortho= 1,2.
For (iii) the parent is a 6-membered ring (cyclohexane); place substituents diametrically.
For (iv) the parent is an 8-C chain (octane). The phrase ``2-(2-chlorophenyl)'' attaches a benzene ring at C2 of the octane chain, and the benzene ring itself carries a Cl at its own C2 (the ring carbon adjacent to the link).
For (vi) the common-name fragment tert-butyl = -C(CH3)3 attaches like any other substituent; sec-butyl in (vii) is -CH(CH3)CH2CH3. Memorise the four C4 substituent fragments (n-, iso-, sec-, tert-) since they recur in CBSE questions.
Sanity-check: re-derive the IUPAC name from each drawn structure to confirm it matches. If the recovered name differs in any locant, your placement is wrong.
Why this matters. Reversibility (name ↔ structure) is a key NCERT exam ask. Practising both directions on the same compound trains the eye. In the 2-mark NCERT format, half marks are for the structure and half for showing the locant assignment; neat structural drawings get full marks even without a re-derivation step.
Structures as drawn in the main solution.
Q 6.4
Which one of the following has the highest dipole moment?
(i) CH2Cl2
(ii) CHCl3
(iii) CCl4
Concept used. The molecular dipole moment (μ⃗) is the vector sum of the individual bond dipoles. In a symmetric molecule, equal and opposite bond dipoles cancel, giving μ⃗=0. In an asymmetric molecule, the cancellation is incomplete, so the molecule has a net dipole. The magnitude depends on (i) the orientation of bond dipoles and (ii) whether C-H contributions reinforce or oppose the C-Cl contributions.
[See diagram in the PDF version]
Identify the geometry. All three molecules are tetrahedral (the central C is sp3). Bond angles are close to 109.5∘.
CCl4. Four identical C-Cl bonds at tetrahedral angles. By symmetry, the four bond dipoles cancel exactly: μ⃗CCl4 = 0 D.
CH2Cl2. Two C-Cl dipoles point ``downwards'' (towards the chlorines), and two C-H dipoles point ``upwards'' (towards the hydrogens, because C is more electronegative than H so the bond dipole points C → H, i.e. outward at H). The two pairs of dipoles point in the same general direction along the bisector axis, so they add. The measured value is μ ≈ 1.60 D.
CHCl3. One C-H dipole and three C-Cl dipoles. The three C-Cl dipoles partially cancel each other; their axial components point opposite to the C-H dipole. The two contributions therefore partially oppose: the net is μ ≈ 1.04 D.
Compare. μ(CH2Cl2) > μ(CHCl3) > μ(CCl4) = 0. So CH2Cl2 has the largest dipole moment.
CH2Cl2 has the highest dipole moment (∼ 1.60 D), because the C-H and C-Cl bond dipoles add constructively, whereas in CHCl3 they partly oppose, and in CCl4 they cancel entirely.
AV
Aditya Verma
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Picture-first. Draw the tetrahedron and add bond-dipole vectors from C toward the more electronegative end of each bond. The vector sum is the molecular dipole.
Vector intuition. Treat each bond dipole as a vector of length bond pointing from the less- to the more-electronegative atom. For symmetric arrangements (tetrahedral, trigonal-planar, linear) where all four bonds are equal, the resultant is zero. The moment a single atom is swapped (say Cl for H), one of the cancelling vectors is replaced by a different vector, and the resultant becomes non-zero. The size of the resultant depends on the angle between the surviving cluster vectors.
CCl4: four equal C-Cl vectors arranged tetrahedrally. The sum of vectors from the centre of a regular tetrahedron to its four vertices is the zero vector. Hence μ = 0 D.
CHCl3: replace one Cl of CCl4 with H. The C-H bond dipole points toward C (since C > H), opposite to the C-Cl dipole that was removed. The single C-H dipole partly opposes the net C-Cl dipole, so the molecule has a small net moment, ∼ 1.04 D.
CH2Cl2: two C-Cl dipoles and two C-H dipoles, with the two halves on opposite faces of the tetrahedron. The vector sums of the two C-Cl dipoles and of the two C-H dipoles point in the same direction, so they add. Net μ ≈ 1.60 D, the largest of the three.
Experimental dipole moments (NCERT Table 6.2): CH3Cl = 1.86 D, CH2Cl2 = 1.60 D, CHCl3 = 1.04 D, CCl4 = 0 D. The trend CH3Cl > CH2Cl2 > CHCl3 > CCl4 is governed by symmetry, not by the count of polar bonds.
Common pitfall to avoid. ``More C-Cl bonds means higher dipole'' is wrong. The dipole of CH3Cl is even larger than CH2Cl2 although it has fewer Cl atoms, because in CH3Cl all three C-H dipoles add constructively with the single C-Cl along the C-Cl axis.
Numerical cross-check. A back-of-envelope estimate for CH2Cl2: each C-Cl bond moment is about 1.5 D; with a Cl–C–Cl angle of ∼ 109.5∘ the resultant of the two C-Cl dipoles is 2 × 1.5 cos(54.7∘) ≈ 1.73 D, then add the two C-H contributions (∼ 0.4 D each, in the same direction) and you reach the observed ∼ 1.6 D.
Why this matters. Counting polar bonds is a beginner's trap. Always sketch the geometry and add vectors. The same vector logic explains why p-dichlorobenzene has μ = 0 while o- and m-isomers do not (Q 6.18), why NH3 and NF3 differ in dipole sign, and why CO2 (μ = 0) and H2O (μ ≠ 0) behave so differently.
CH2Cl2 has the highest dipole moment.
Q 6.5
A hydrocarbon C5H10 does not react with chlorine in dark but gives a single monochloro compound C5H9Cl in bright sunlight. Identify the hydrocarbon.
Concept used. The degree of unsaturation Ω for a hydrocarbon C_n H_m is Ω = 2n + 2 - m2. Ω counts the total number of rings + double bonds + triple bonds (each double = 1, each triple = 2, each ring = 1). For C5H10, Ω = (10 + 2 - 10)/2 = 1, so the molecule has exactly one ring or one C=C.
Why ``no reaction in the dark'' rules out an alkene
Alkenes react with Cl2 in the dark by electrophilic addition across the C=C (no light needed). If a C5H10 compound does not react with Cl2 in the dark, it cannot contain a C=C. So Ω = 1 must come from a ring: the compound is a cycloalkane.
Compute Ω for C5H10: Ω = (2· 5 + 2 - 10)/2 = 2/2 = 1. One ring or one double bond.
``Dark + no reaction'' rules out a C=C. The compound is a cyclopentane (C5H10, one ring).
Alkanes and cycloalkanes react with Cl2 in sunlight by a free-radical chain: initiation Cl2 hν 2 Cl., propagation Cl. + R-H -> H-Cl + R. and R. + Cl2 -> R-Cl + Cl..
``One single monochloride'' means all the C-H bonds in the molecule are equivalent. Among the C5H10 cycloalkanes, only cyclopentane has every hydrogen equivalent (a 5-membered ring with all -CH2- groups chemically identical).
Verify: chlorination of cyclopentane gives only chlorocyclopentane: C5H10 + Cl2 hν C5H9Cl + HCl.
[See diagram in the PDF version]
The hydrocarbon is cyclopentane, C5H10. Its monochloride is chlorocyclopentane.
AS
Arjun Singh
M.Sc Chemistry, IIT Kanpur
Verified Expert
Strategic angle. Two clues, two filters: ``no dark reaction with Cl2'' eliminates alkenes; ``single monochloride'' demands all-equivalent hydrogens.
Symmetry counting in detail. To check whether all H's in a cycloalkane are equivalent, draw the ring and mark each ring carbon. Use the ring's rotational and mirror symmetry to test if any two H's can be carried into each other by a symmetry operation. For cyclopentane, the D5h symmetry has C5 rotational axis: any ring CH2 can be carried onto any other by a 72∘ rotation, and the two H's on each CH2 are related by the in-plane mirror. All 10 H's are equivalent.
Filter 1: ``no reaction in dark with Cl2'' means no C=C. Since the formula C5H10 has one degree of unsaturation, that unsaturation must be a ring.
Filter 2: ``single monochloride'' means there is only one kind of C-H. For five-carbon cycloalkanes, the candidates are cyclopentane (all 10 H's equivalent), methylcyclobutane (4 kinds of H), and ethylcyclopropane (5 kinds of H). Only cyclopentane survives.
Reaction is photochemical (Cl2 hν 2 Cl.); the Cl^. radical abstracts an H to give a 5-membered radical, which reacts with Cl2 to give the monochloride. The overall stoichiometry is C5H10 + Cl2 -> C5H9Cl + HCl.
Confirm by counting hydrogens: cyclopentane has 10 equivalent C-H bonds, so abstracting any of them gives the same radical and the same product.
Common pitfall to avoid. Don't forget that methylcyclobutane and ethylcyclopropane also satisfy C5H10 and have one ring (so Ω = 1, no C=C). It is the ``single monochloride'' clue that rules them out, not the dark-test clue.
Concept linkage. The same reasoning identifies neopentane (2,2-dimethylpropane, C5H12) from radical bromination giving a single monobromide; both compounds have Td-related symmetry that puts all C–H's in one equivalence class.
Why this matters. Substitution-selectivity problems on hydrocarbons reduce to symmetry counting. Practice on C4H10 (only n-butane and isobutane), C5H12 (the three pentanes), and C5H10. In JEE-Mains the question is often phrased ``which hydrocarbon gives only one monobromide?'' — the answer is always the most symmetric one.
Cyclopentane.
Q 6.6
Write the isomers of the compound having formula C4H9Br.
Concept used. ``Isomers of C4H9Br'' means all structures consistent with the molecular formula. We enumerate the carbon skeletons of C4H10 (namely n-butane and isobutane) and substitute one H by Br in every non-equivalent position.
n-Butane skeleton CH3-CH2-CH2-CH3: the two terminal CH3 groups are equivalent (one kind of H); the two internal CH2 groups are equivalent (a second kind of H). Substituting gives:
[leftmargin=*]
1-Bromobutane: CH3-CH2-CH2-CH2Br (primary, 1∘).
2-Bromobutane: CH3-CH2-CH(Br)-CH3 (secondary, 2∘). This carbon has four different groups (H, Br, CH3, CH2CH3), so it is a stereocentre; the compound exists as a pair of enantiomers (R and S).
Isobutane skeleton (CH3)3CH: the three CH3 groups are equivalent (one kind of H); the lone central CH is a second kind of H. Substituting gives:
Therefore there are four constitutional isomers of C4H9Br, namely 1-bromobutane, 2-bromobutane, 1-bromo-2-methylpropane and 2-bromo-2-methylpropane. Counting the enantiomeric pair of 2-bromobutane, five total stereoisomers exist.
0.95!%
[See diagram in the PDF version]
Four constitutional isomers: 1-bromobutane, 2-bromobutane, 1-bromo-2-methylpropane and 2-bromo-2-methylpropane. 2-Bromobutane itself comes as a pair of enantiomers (R and S), giving five total stereoisomers.
PN
Pranav Nair
M.Sc Chemistry, IIT Kanpur
Verified Expert
Structural observation. C4H9Br is saturated with no rings: enumerate by carbon skeleton, then by H-replacement position.
Two-step enumeration recipe. For any C_n H_2n+1 X: (1) list all carbon skeletons of C_n H_2n+2; for n = 4 that is 2 (n-butane, isobutane); for n = 5 that is 3 (n-pentane, isopentane, neopentane). (2) On each skeleton, identify each set of equivalent H's and substitute one at a time. The number of constitutional isomers equals the sum of inequivalent H sets across skeletons.
For C4 the two carbon skeletons are n-butane (linear) and isobutane (branched).
In n-butane, two non-equivalent H positions exist (terminal CH3 and internal CH2): 1-bromobutane and 2-bromobutane.
In isobutane, two non-equivalent H positions exist (terminal CH3 on any of the three equivalent arms, and the central CH): 1-bromo-2-methylpropane and 2-bromo-2-methylpropane.
2-Bromobutane has a stereocentre at C2 (four different groups: H, Br, methyl, ethyl): it is chiral and exists as a pair of enantiomers.
Identity check. Confirm each isomer by counting H's: 1-Br-butane = C4H9Br (3+2+2+2 H + 0 on Br C = 9 H, ); 2-Br-butane = 3+2+1+3 = 9 H ; isobutyl bromide = 6+1+2 = 9 H ; t-butyl bromide = 9 + 0 = 9 H . All match the molecular formula.
Class of each. 1-Br-butane (1∘), 2-Br-butane (2∘), isobutyl bromide (1∘), t-butyl bromide (3∘). The two primaries give slow SN1; the secondary mixes SN1/SN2; the tertiary gives only SN1 (and E1).
Why this matters. Counting isomers is a common JEE/CBSE Q. The recipe (skeletons × non-equivalent H sites) generalises to any C_n H_2n+1 X. The same enumeration template also produces all isomers of C5H12 (3), C5H11Br (8), and other simple haloalkane problems.
Four constitutional isomers; 2-bromobutane adds an R/S pair for a total of five stereoisomers.
Q 6.7
Write the equations for the preparation of 1-iodobutane from
(i) 1-butanol
(ii) 1-chlorobutane
(iii) but-1-ene.
Concept used. 1-Iodobutane (CH3CH2CH2CH2I) can be prepared from each precursor by a different route:
[leftmargin=*]
From an alcohol: convert -OH to -I using red phosphorus and iodine, which generates HI in situ.
From a chloroalkane: a Finkelstein reaction in dry acetone: NaI dissolves in acetone whereas NaCl does not. Precipitation of NaCl drives the equilibrium toward the iodide.
From an alkene: anti-Markovnikov addition of HBr with peroxide (Kharasch effect) gives 1-bromobutane, which is converted to 1-iodobutane by Finkelstein exchange. Peroxide effect does not work for HI, so we cannot go directly from but-1-ene to 1-iodobutane.
(i) From 1-butanol. CH3CH2CH2CH2OH + HI Δ CH3CH2CH2CH2I + H2O. The HI is generated from red P and I2: 3 CH3CH2CH2CH2OH + P + 3/2 I2 -> 3 CH3CH2CH2CH2I + H3PO3. Mechanism: HI protonates the alcohol O to give the oxonium R-OH2+, which is attacked by I- in an SN2 step (backside attack on C, water leaves).
(ii) From 1-chlorobutane (Finkelstein). CH3CH2CH2CH2Cl + NaI dry acetone CH3CH2CH2CH2I + NaCl(s). Mechanism is SN2: I- attacks the primary C-Cl carbon from the back, displacing Cl-. The reaction is driven by precipitation of NaCl (insoluble in acetone).
(iii) From but-1-ene. Two-step sequence. Step (a): anti-Markovnikov addition of HBr with peroxide (Kharasch effect): CH3CH2CH=CH2 + HBr 4ptperoxide CH3CH2CH2CH2Br. Mechanism: peroxide initiates Br^., which adds to the terminal C (giving the more stable secondary radical); chain transfer with HBr gives 1-bromobutane. Step (b): Finkelstein exchange: CH3CH2CH2CH2Br + NaI dry acetone CH3CH2CH2CH2I + NaBr(s).
0.95!%
[See diagram in the PDF version]
(i) CH3CH2CH2CH2OH HI, Δ CH3CH2CH2CH2I; (ii) CH3CH2CH2CH2Cl + NaI acetone CH3CH2CH2CH2I + NaCl; (iii) CH3CH2CH=CH2 HBr/peroxide CH3CH2CH2CH2Br NaI, acetone CH3CH2CH2CH2I.
KR
Karan Reddy
M.Tech Chemical Engineering, IIT Delhi
Verified Expert
Strategic angle. Aim at the same end product CH3CH2CH2CH2I from three different starting points: a hydroxyl, a chloride, and an alkene.
Alternative reagents (board-friendly substitutes). For (i), SOCl2 then NaI/acetone is a longer but equally valid two-step route to the chloride first, then Finkelstein. PCl5 or PCl3 would give the chloride from the alcohol if you prefer working through 1-chlorobutane. NCERT explicitly mentions P + I2/red P for direct conversion to the iodide.
For 1-butanol, the practical lab reagent is P + I2, which yields HI in situ. The overall stoichiometry is 3 ROH + P + 3/2 I2 -> 3 RI + H3PO3. Each ROH gets attacked by HI in two steps: protonate OH, then SN2 by I-.
For 1-chlorobutane, Finkelstein. The driving force is differential solubility: NaI is soluble in dry acetone, but the product NaCl is not. Le Chatelier pulls the equilibrium fully to the right.
For but-1-ene, do not try direct HI addition (it gives 2-iodobutane, not the desired 1-iodobutane). Instead, Kharasch-add HBr/peroxide to get 1-bromobutane (anti-Markovnikov), then Finkelstein to swap Br for I.
Confirm regioselectivity in step (iii): Br^. adds to the terminal CH2 (less substituted carbon) because that puts the radical on the more substituted (more stable) secondary carbon.
Why HI peroxide doesn't work. The H–I bond (∼ 297 kJ/mol) is much weaker than C–I (∼ 234 kJ/mol), so the chain-transfer step R. + HI -> R-H + I. is exothermic (favourable) but the alkene-addition step I. + C=C -> I-C-C. is too endothermic to sustain a chain. So peroxide effect runs cleanly only for HBr, not HI or HCl.
Concept linkage. This question previews retrosynthesis: every chain-modifying disconnection in organic chemistry comes back to one of these named transformations (alcohol → halide → Finkelstein, alkene → Markovnikov/anti-Markovnikov HX).
Why this matters. These three routes summarise the three canonical disconnections to a primary alkyl iodide. In a CBSE board question, examiners explicitly state ``write three different conversions'' to test whether students can pick the correct reagent for each starting material.
(i) P/I2 on 1-butanol; (ii) NaI/acetone on 1-chlorobutane; (iii) HBr/peroxide on but-1-ene, then NaI/acetone.
Q 6.8
What are ambident nucleophiles? Explain with an example.
Concept used. A nucleophile is a species with a lone pair (or π-electrons) that attacks an electrophilic centre. An ambident nucleophile (Latin ambi- = both, dent = tooth) has two different reactive sites; either can serve as the donor. Which site attacks depends on:
[leftmargin=*,itemsep=1pt]
the hardness/softness of the nucleophile and electrophile (HSAB principle: hard prefers hard, soft prefers soft);
the mechanism (SN2 vs SN1);
the solvent, the temperature, and the kinetic-vs- thermodynamic control.
The classical example: cyanide ion CN-
CN- has lone pairs on both C and N. Attack via C gives an alkyl cyanide (nitrile) R-C#N; attack via N gives an alkyl isocyanide R-N#C.
Cyanide ion CN- has lone pairs on both C and N (resonance pushes negative charge between the two ends). Both ends are nucleophilic.
With KCN (ionic salt, polar solvent), the C end is the more nucleophilic end and attacks the alkyl halide: R-X + K+CN- -> R-C#N + K+X- (alkyl cyanide).
With AgCN (more covalent salt with the Ag-C bond locking the C lone pair), the lone pair on N is more available; the N end attacks: R-X + Ag-C#N -> R-N#C + AgX(s) (alkyl isocyanide).
Other ambident nucleophiles: nitrite NO2- (gives nitro R-NO2 via N, or nitrite ester R-O-N=O via O); thiocyanate SCN- (gives R-SCN or R-NCS); enolate (C-alkylation or O-alkylation).
0.95!%
[See diagram in the PDF version]
An ambident nucleophile has two different reactive sites. Example: cyanide ion CN- attacks via C (giving R-CN) when paired with KCN, and via N (giving R-NC) when paired with AgCN.
VB
Vivaan Bhat
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Structural observation. Whenever you see a nucleophile written with delocalised charge over two atoms (O/N, C/N, S/N), suspect ambident behaviour.
[leftmargin=*,itemsep=1pt]
CN-: C or N.
NO2-: N or O.
SCN-: S or N.
HSAB applied carefully. Hard nucleophiles (small, electronegative, weakly polarisable) prefer hard electrophiles (small, positive). Soft nucleophiles (large, polarisable, often neutral) prefer soft electrophiles (large, partial positive). In CN-, the N end is harder (more electronegative); the C end is softer (more polarisable). For an alkyl halide with δ+ on a soft carbon, C attack dominates. For early (SN1-like) transition states with a more localised charge, the N attack route opens up.
For CN- with KCN, the ionic K–C bond is fully ionised; the C lone pair is free and attacks R-X in SN2, producing the nitrile.
For CN- with AgCN, Ag forms a covalent Ag-C bond, locking the C lone pair and freeing the N lone pair to attack, producing the isocyanide.
Selectivity also depends on solvent: protic solvents hydrogen-bond the more electronegative end, slowing it down and shifting attack to the other end.
Exam-tip framing. In NCERT exercise answers, always cite the example as CN- with KCN vs. AgCN — this is the textbook canonical pair and is the one CBSE markers expect. Mention NO2-, SCN- as additional examples for the 3-mark variant.
Concept linkage. Ambident nucleophiles appear again in chapter 7 (Alcohols, Phenols, Ethers): the phenoxide C6H5O^- is ambident between O and the ortho/para ring carbons, leading to Kolbe–Schmidt and Reimer–Tiemann reactions where C- rather than O-attack dominates.
Why this matters. Same starting material plus the same nucleophile, two different products. NCERT exam writes this as a one-mark or short-answer question. The deeper idea — that delocalised charge enables more than one nucleophilic site — recurs through enolates, phenoxides, carboxylates and is foundational for much of the rest of class 12 organic chemistry.
Same conclusion: CN- (and NO2-, SCN-) is ambident; R-X + KCN -> R-CN but R-X + AgCN -> R-NC.
Q 6.9
Which compound in each of the following pairs will react faster in SN2 reaction with OH-?
(i) CH3Br or CH3I
(ii) (CH3)3CCl or CH3Cl
Concept used. The SN2 (bimolecular nucleophilic substitution) rate law is rate = k [R-X] [Nu-]. The nucleophile attacks the carbon from the side opposite to the leaving group (backside attack), through a single trigonal-bipyramidal transition state in which the C is partially bonded to both Nu and X. The reaction is fastest when
[leftmargin=*,itemsep=1pt]
the leaving group is a good leaving group (weakest base, best polarisable: I^- > Br^- > Cl^- > F^-);
the substrate is sterically unhindered (methyl >1∘>2∘>3∘);
the nucleophile is strong and the solvent is polar aprotic.
[See diagram in the PDF version]
(i) CH3Br vs CH3I. The two have the same carbon framework (methyl, no steric difference), so the rate is decided by the leaving group. I^- is larger and more polarisable than Br^-, and is a weaker base (HI is a stronger acid than HBr), so I^- is a better leaving group. The C-I bond (∼ 234 pm) is also weaker than the C-Br bond (∼ 194 pm), so it breaks more easily. Therefore CH3I reacts faster than CH3Br in SN2.
(ii) (CH3)3CCl vs CH3Cl. The leaving group is Cl^- in both, so the rate is decided by steric crowding at the electrophilic C. CH3Cl is methyl (no β-substituents), so the backside is wide open. (CH3)3CCl is tertiary, with three CH3 groups crowding the backside; the transition state is destabilised by steric repulsion and the rate is essentially zero. Therefore CH3Cl reacts much faster than (CH3)3CCl in SN2.
(i) CH3I reacts faster (better leaving group). (ii) CH3Cl reacts faster (no steric hindrance at C).
AJ
Aanya Joshi
M.Sc Chemistry, IIT Kanpur
Verified Expert
Strategic angle. Two single-question filters: leaving-group ability for (i), steric accessibility for (ii).
Quantitative perspective. Relative SN2 rates with OH- (taking CH3I = 1): CH3I ≈ 1.0, CH3Br ≈ 0.02 — a ∼ 50× rate difference. For the steric pair: CH3Cl/(CH3)3CCl ≈ 106 — six orders of magnitude. These ratios underline how different the two controlling factors are in magnitude.
Leaving-group ability for halides follows I^- > Br^- > Cl^- > F^-. The rule of thumb: the weaker the conjugate base, the better the leaving group, and HI (pKa ≈ -10) is much more acidic than HBr (pKa ≈ -9).
For (i), same carbon, different X: CH3I wins because the C–I bond is weaker and I^- is a better leaving group.
Steric hindrance to backside attack scales as 3∘ ≫ 2∘ > 1∘ ≈ methyl. The SN2 transition state has a crowded 5-coordinate C; more substituents means worse crowding.
For (ii), same X, very different steric situation: methyl chloride is essentially fully exposed; tert-butyl chloride has three methyls blocking the backside. Methyl chloride wins by orders of magnitude.
Alternative perspective: bond strength. The C–X bond strengths (kJ/mol) are roughly C–F (485), C–Cl (327), C–Br (285), C–I (213). Weaker bond = easier breaking = better leaving group. This trend reinforces (does not contradict) the basicity argument.
Concept linkage. The same orderings carry into E2 eliminations (I- fastest, F- slowest leaving) and into haloarene reactivity (where F on benzene is actually fastest in the addition-elimination mechanism of nucleophilic aromatic substitution — the opposite of the SN2 trend! See chapter 6 sections on aryl halides).
Why this matters. A two-line answer per pair is enough for NCERT marking; the underlying reasoning (LG ability + steric access) is universal across SN2 questions. CBSE markers expect both the order and one reason (LG ability OR steric) for full marks.
(i) CH3I; (ii) CH3Cl.
Q 6.10
Predict all the alkenes that would be formed by dehydrohalogenation of the following halides with sodium ethoxide in ethanol and identify the major alkene:
(i) 1-Bromo-1-methylcyclohexane
(ii) 2-Chloro-2-methylbutane
(iii) 2,2,3-Trimethyl-3-bromopentane.
Concept used.Dehydrohalogenation (also called β-elimination or E2) removes one H from a β-carbon (the carbon next to the one bearing X) and X from the α-carbon, giving an alkene. Strong bases like sodium ethoxide (C2H5O-Na+) favour E2 over substitution. According to Saytzeff's rule, the major alkene is the most substituted one (more alkyl groups stabilise the C=C via hyperconjugation).
(i) 1-Bromo-1-methylcyclohexane. The Br sits on a ring carbon that also carries a CH3. β-positions: two ring carbons adjacent to the C-Br, both -CH2- (equivalent by symmetry), and the methyl group (which contributes β-H's, giving an exocyclic =CH2).
Possible alkenes:
[leftmargin=*]
Loss of H from a ring CH2: gives 1-methylcyclohex-1-ene (trisubstituted).
Loss of H from the methyl: gives methylenecyclohexane (disubstituted).
Saytzeff: trisubstituted > disubstituted; the major product is 1-methylcyclohex-1-ene.
(ii) 2-Chloro-2-methylbutane. The Cl sits on C2 of the chain CH3-C(Cl)(CH3)-CH2-CH3. β-carbons: C1 (a CH3, terminal), C3 (a CH2, internal), and the methyl substituent on C2.
Possible alkenes:
[leftmargin=*]
Loss of H from C3 CH2: gives 2-methylbut-2-ene (trisubstituted).
Loss of H from C1 CH3 or from the methyl substituent: gives 2-methylbut-1-ene (disubstituted).
Major: 2-methylbut-2-ene.
(iii) 2,2,3-Trimethyl-3-bromopentane. (CH3)3C-C(Br)(CH3)-CH2-CH3. The Br is on C3. β-carbons: C2 (a quaternary C(CH3)3, no H, so no elimination there), C4 (a CH2), and the methyl substituent on C3.
Possible alkenes:
[leftmargin=*]
Loss of H from C4: gives 3,4,4-trimethylpent-2-ene (trisubstituted).
Loss of H from the methyl substituent on C3: gives 2,3,3-trimethylpent-1-ene (disubstituted).
Strategic angle. Identify every β-hydrogen on a distinct β-carbon, draw the alkene formed by removing each in turn, then rank by alkene substitution (Saytzeff).
Mechanism perspective.E2 is concerted: the C–H bond, the C–C bond and the C–X bond all move together in one transition state. The base (EtO-) abstracts H as X- leaves; the two carbons rehybridise from sp3 to sp2. A key geometric requirement is that the H and X sit antiperiplanar (dihedral ≈ 180∘). In open-chain systems this is achieved by rotation; in cyclic systems it constrains which H can be removed (this matters for ring systems with conformational locks like methylcyclohexane derivatives).
For (i), the two ring CH2 are equivalent: they give one product (1-methylcyclohex-1-ene). The methyl β-H gives methylenecyclohexane. Trisubstituted vs disubstituted; major is the trisubstituted ring alkene.
For (ii), the C1 methyl and the C2-methyl substituent give the same terminal alkene 2-methylbut-1-ene; the C3 CH2 gives 2-methylbut-2-ene. Major: 2-methylbut-2-ene (trisubstituted).
For (iii), the quaternary C2 has no β-H. The C4 CH2 gives an internal trisubstituted alkene; the methyl on C3 gives a terminal disubstituted alkene. Major: 3,4,4-trimethylpent-2-ene.
Count the substitution: trisubstituted has 3 alkyl groups on C=C; disubstituted has 2. Saytzeff says ``more''.
Numerical check (alkene stability). Heats of hydrogenation (kJ/mol, smaller = more stable alkene): monosubstituted ≈ -126, disubstituted ≈ -116, trisubstituted ≈ -113, tetrasubstituted ≈ -111. The ∼ 5 kJ/mol difference per added substituent is the energetic basis of Saytzeff's rule.
Common pitfall in (i). Students sometimes count only the ring CH2 groups and forget the methyl. The methyl is itself a β-position (carbons one bond from C-Br), and removing one of its H's gives the terminal alkene methylenecyclohexane. Both products are valid; only the relative amount differs.
Concept linkage. Saytzeff governs E1 as well (carbocation → most substituted alkene). When the base is bulky (like t-BuO-, hindered amines), Hofmann elimination kicks in and the least substituted alkene becomes major because the bulky base can only reach the least crowded β-H.
Why this matters. Saytzeff vs Hofmann is a one-mark selection problem in NCERT/JEE. The recipe: list β-H sites, count alkene substitution, pick the most substituted (with a small base). For 3-mark questions, also draw the alkenes' structural formulae and label the substitution pattern.
Same Saytzeff majors as in main solution.
Q 6.11
How will you bring about the following conversions?
(i) Ethanol to but-1-yne; (ii) Ethane to bromoethene; (iii) Propene to 1-nitropropane; (iv) Toluene to benzyl alcohol; (v) Propene to propyne; (vi) Ethanol to ethyl fluoride; (vii) Bromomethane to propanone; (viii) But-1-ene to but-2-ene; (ix) 1-Chlorobutane to n-octane; (x) Benzene to biphenyl.
Concept used. Each conversion uses a small set of named transformations: halogenation, elimination, nucleophilic substitution, Wurtz coupling, Grignard chemistry, etc.
(vi) Ethanol → ethyl fluoride. CH3CH2OH HBr or PBr3 CH3CH2Br Hg2F2 or AgF CH3CH2F.
(vii) Bromomethane → propanone. CH3Br KCN CH3CN CH3MgBr; H3O+ CH3-CO-CH3. (Grignard attacks the nitrile carbon; the imine salt hydrolyses to a ketone.)
Ten conversions answered above with full reagent sequences and stepwise equations.
DC
Diya Chatterjee
B.Tech Chemical Engineering, IIT Bombay
Verified Expert
Strategic angle. Each conversion is a retrosynthesis. Walk backward from the target to the starting material; identify the disconnection(s); pick standard reagents.
For (i) but-1-yne, the disconnection C-C between an ethyl and the terminal alkyne gives ethyne + CH3CH2Br. Ethyne is built from ethanol via ethylene, dibromide, double elimination.
For (ii) bromoethene, the disconnection at C=C-Br suggests CHBr=CH2 from CH2Br-CH2Br by single elimination. Build the dibromide from ethylene; ethylene from bromoethane (radical bromination of ethane).
For (iii), R-NO2 from R-Br + AgNO2. The R-Br is from propene by Kharasch addition of HBr with peroxide.
For (iv), Ph-CH2OH from Ph-CH2Cl via hydrolysis. Get Ph-CH2Cl from toluene by photochemical side-chain chlorination.
For (v), double dehydrohalogenation of a vicinal dibromide. Build the dibromide from propene and Br2.
For (vi), since HF cannot substitute -OH directly, route through bromide and swap to fluoride with AgF.
For (vii), nitrile-to-ketone sequence: CH3Br -> CH3CN -> CH3-CO-CH3 via Grignard addition and hydrolysis.
For (ix), Wurtz couples two molecules of R-Cl with Na in dry ether to give the R-R dimer.
For (x), Wurtz–Fittig couples two Ph-Br with Na in dry ether to give biphenyl.
Why this matters. These ten conversions span the major named reactions of this chapter (Wurtz, Wurtz–Fittig, Finkelstein, Saytzeff, Kharasch, Grignard). Multi-mark CBSE questions sometimes recombine 4–5 of them as a single ``starting from X, give Y'' chain; practising all ten in advance puts you in a position to recognise any sub-chain instantly.
Concept linkage. Notice that conversions (i), (ii), (iv) all use a haloalkane intermediate; (v) uses two elimination steps in sequence. Most multistep retrosynthesis in class 12 organic chemistry passes through one or more haloalkanes — they are the chapter's pivot point because every C–X bond breaks (substitution) or its β-H leaves (elimination) under known conditions.
Same ten sequences with same reagents.
Q 6.12
Explain why
(i) the dipole moment of chlorobenzene is lower than that of cyclohexyl chloride?
(ii) alkyl halides, though polar, are immiscible with water?
(iii) Grignard reagents should be prepared under anhydrous conditions?
Concept used.
[leftmargin=*,itemsep=1pt]
Dipole moment μ = q · d depends on the partial charge q and the bond length d. Hybridisation and π-donation affect q; bond length affects d.
Miscibility depends on whether mixing breaks more strong H-bonds than it forms.
A Grignard reagent (R-Mg-X) is a strongly polar organometallic with δ- on C, behaving as a strong base/nucleophile.
(i) μ(chlorobenzene) < μ(cyclohexyl chloride). Two reasons:
[label=(),leftmargin=*]
Hybridisation of carbon: in chlorobenzene, the C attached to Cl is sp2 (s-character ∼ 33%); in cyclohexyl chloride, sp3 (∼ 25%). Higher s-character means a more electronegative C, smaller electronegativity difference with Cl, and a smaller μ for chlorobenzene.
Resonance (+M donation): in chlorobenzene, lone pairs on Cl delocalise into the ring, giving Cl partial positive character and the ring partial negative. This opposes the C-Cl σ dipole, reducing μ.
Combined: C6H5Cl ≈ 1.69 D, C6H11Cl ≈ 2.05 D.
(ii) Alkyl halides are immiscible with water. Water dissolves a solute only if the energy released by forming solute–water attractions (∼H-bonds) compensates for breaking water–water H-bonds. Alkyl halides cannot donate H-bonds (no O-H or N-H) and accept very weakly. Mixing them into water would require breaking a network of strong water–water H-bonds (each ∼ 20 kJ/mol) in exchange for weak van der Waals/halide dipole interactions. The free-energy change is positive, so the two phases stay separate.
(iii) Anhydrous conditions for Grignard. The C-Mg bond in R-Mg-X is strongly polarised: Rδ--Mgδ+, with R acting as a carbanion equivalent. Water has an acidic O-H (pKa ≈ 15.7); the conjugate acid of R- (which is R-H) has pKa ≈ 50. So R- is ∼ 35 pK units more basic than OH-, and the deprotonation is essentially irreversible: R-Mg-X + H2O -> R-H + Mg(OH)X. The Grignard is destroyed before it can react with the intended electrophile.
(i) sp2 hybridisation + resonance reduce μ of chlorobenzene below that of cyclohexyl chloride; (ii) no H-bonding from alkyl halides to water; mixing demands breaking water's own H-bond network; (iii) Grignard reagent is a strong base toward water and is destroyed instantly by moisture.
KP
Krishna Pillai
M.Sc Chemistry, IIT Kanpur
Verified Expert
Strategic angle. Three short ``why?'' questions, three named effects: hybridisation + resonance; H-bond mismatch; acid–base reactivity.
Diagram thinking for (i). Sketch chlorobenzene with the Cl lone pair drawn as an arrow into the ring (the +M resonance contributor); the partial negative on the ring carbons opposes the C-Cl dipole. Add the C-Cl bond-length annotation: ∼ 169 pm (chlorobenzene) vs ∼ 177 pm (cyclohexyl chloride). Both shorter bond and weaker partial charge reduce μ = q · d.
For (i), pair the two factors. Higher s-character at the sp2 ring C makes it more electronegative; the C-Cl bond is also shorter (∼ 169 pm vs ∼ 177 pm for sp3). Combined with +M donation from Cl, the net dipole is reduced.
For (ii), break down miscibility into Δ Hsoln and Δ Ssoln. The positive Δ H from breaking water H-bonds dominates over the small ion–dipole attractions an alkyl halide can offer. Two phases is the lower-energy state.
For (iii), use a pKa argument. The carbanion derived from a Grignard is the conjugate base of an alkane (pKa ≈ 50). Water (pKa = 15.7) is a much stronger acid. The protonation lies ∼ 1034 to the right toward R-H.
Numerical reinforcement for (i). Even though chlorobenzene has higher s-character at C (so the C-Cl bond is more polar in some sense), the resonance donation reduces the partial negative on Cl. Both effects together drop μ from ∼ 2.05 D (cyclohexyl chloride) to ∼ 1.69 D (chlorobenzene) — an 18% decrease.
Practical note on (iii). The Grignard rule ``no moisture'' extends to apparatus: glassware must be flame-dried; solvents (dry ether or THF) must be distilled over Na/benzophenone; Mg turnings should be activated with a tiny crystal of I2 to remove surface oxide. A single drop of water can quench a Grignard run that took hours to set up.
Concept linkage. The same protic-incompatibility of Grignards extends to alcohols (ROH also kills Grignards), amines (R2NH), terminal alkynes (HC#CR), and acids — any O-H, N-H, S-H, or terminal sp-CH with pKa < 50 will protonate R-MgX.
Why this matters. Each of these three explanations is a self-contained one-mark answer NCERT loves to ask. The reasoning templates (resonance + hybridisation; energetics of solvation; pKa logic) recur in chapters on Alcohols, Amines and Carbonyls.
Same three explanations, with the quantitative arguments above.
Q 6.13
Give the uses of freon 12, DDT, carbon tetrachloride and iodoform.
Concept used. Polyhalogenated compounds find use because of their high density, low flammability, low chemical reactivity, high boiling points (chlorinated solvents); insecticidal action (chlorinated aromatics); pharmaceutical antiseptic action (CHI3); and refrigerant properties (volatile fluorochlorocarbons).
Freon-12 (CCl2F2, dichlorodifluoromethane).
[leftmargin=*]
Refrigerant in domestic and industrial refrigeration and air-conditioning.
Aerosol propellant in spray cans.
Phased out under the Montreal Protocol; CFCs deplete stratospheric ozone.
DDT (p,p'-dichlorodiphenyltrichloroethane).
[leftmargin=*]
First synthetic chlorinated insecticide; effective against malaria-carrying mosquitoes and lice.
Banned for agriculture in many countries because of bioaccumulation, persistence, and toxicity to non-target species.
Carbon tetrachloride (CCl4).
[leftmargin=*]
Industrial solvent for fats, oils, resins.
Earlier used in fire extinguishers (Pyrene-type); discontinued because hot CCl4 can generate phosgene (COCl2, toxic).
Feedstock for CCl2F2 (freon-12) synthesis.
Iodoform (CHI3).
[leftmargin=*]
Antiseptic for dressing wounds (the antiseptic action comes from liberated free I2, not from CHI3 itself).
Largely replaced by modern antiseptics because of its unpleasant smell and irritation potential.
Freon-12: refrigerant + aerosol propellant; DDT: insecticide (now restricted); CCl4: industrial solvent, formerly fire extinguisher; CHI3: antiseptic for wound dressings.
ID
Ishaan Desai
M.Sc Chemistry, IIT Kanpur
Verified Expert
Quick reading. Four common polyhalogen compounds, four common uses, four corresponding environmental footnotes.
Structure → property logic. Each compound's utility comes from a structural feature you can read off the formula: freon-12 (CCl2F2) has small molecular mass and zero net H-bonding → low b.p., volatile, good refrigerant. CCl4 is heavy and symmetric (zero dipole, no H-bond) → dense solvent for non-polar oils. CHI3 is heavy (low volatility) yet hydrolytically unstable → slow I2 release. DDT's chlorinated aromatic core is lipophilic and persistent → long-lasting insecticide.
DDT: insecticide, especially anti-malarial vector control. Property used: lipophilic, neurotoxic to insects.
CCl4: solvent for fats/oils, formerly a fire extinguisher. Property used: dense, non-flammable.
CHI3: antiseptic (releases I2, which is the actual germicide). Property used: slow I2 release.
Environmental caveats (board-asked). CFCs catalytically destroy stratospheric O3 (Rowland–Molina mechanism). DDT bioaccumulates (lipid-soluble, long biological half-life). CCl4 is hepatotoxic and forms COCl2 (phosgene) in heat. CHI3 causes skin irritation and has an unpleasant odour.
Replacements (modern chemistry). Freon-12 has been replaced by HFCs (e.g. CH2FCF3) and HFOs that contain no Cl (so no ozone depletion). CCl4 in fire-fighting has been replaced by CO2 and dry chemical extinguishers. DDT use is heavily restricted but the WHO still permits indoor residual spraying against malaria mosquitoes in some regions.
Why this matters. NCERT pairs the use with an environmental caveat in each case; expect a one-mark question or a part of a multi-mark question asking ``write one use and one disadvantage''. The Montreal Protocol (1987) and Stockholm Convention (2001) are international agreements students should be aware of for the 2-mark ``mention one international protocol'' extension.
Same uses as the main solution.
Q 6.14
Write the structure of the major organic product in each of the following reactions:
(i) CH3CH2CH2Cl + NaI ->
(ii) (CH3)3CBr + KOH ->
(iii) CH3CH(Br)CH2CH3 + NaOH ->
(iv) CH3CH2Br + KCN ->
(v) C6H5ONa + C2H5Cl ->
(vi) CH3CH2CH2OH + SOCl2 ->
(vii) CH3CH2CH=CH2 + HBr ->
(viii) CH3CH=C(CH3)2 + HBr ->
Concept used.
[leftmargin=*,itemsep=1pt]
Finkelstein exchange: R-Cl + NaI acetone R-I + NaCl(s).
Strategic angle. Eight one-line transformations, each matching a named reaction. Identify the reaction class, write the product directly.
Reaction-class index. Build a mental table: (i) Finkelstein (halide exchange); (ii) E2 (tertiary + alcoholic base); (iii) SN (secondary + aqueous base); (iv) SN with ambident nucleophile (CN- via C end); (v) Williamson ether synthesis; (vi) SOCl2 clean chlorination; (vii), (viii) Markovnikov HX addition. Reading the reagent first makes the answer ``write itself''.
(i) Finkelstein in dry acetone; primary chloride → iodide.
(ii) 3∘ bromide + alcoholic KOH →E2; only one Saytzeff alkene possible.
(iii) 2∘ bromide + aqueous NaOH → substitution to give the secondary alcohol.
(iv) KCN's C end attacks primary bromide → nitrile.
(vi) SOCl2 converts 1∘ alcohol to 1∘ chloride; gaseous by-products.
(vii) Markovnikov HBr addition to but-1-ene; Br to the more substituted C.
(viii) Markovnikov HBr addition to 2-methylbut-2-ene; Br to the C with two methyl substituents.
Common pitfall in (ii). A tertiary halide with aqueous KOH would also give some elimination because of the substrate's steric crowding; the textbook answer focuses on alcoholic KOH where the elimination channel dominates cleanly. Always read the solvent specification.
Why SOCl2 is preferred over HCl/PCl5 in (vi). The by-products are gases (SO2, HCl) which escape; no separation step needed, and the product is high-purity alkyl chloride. PCl5 leaves POCl3 as a liquid contaminant.
Why this matters. A perfect drill on SN2 vs E2 vs addition, plus the Williamson and SOCl2 shortcuts. Many of these reactions are the building blocks for the multistep conversions in Q 6.11 and Q 6.19, so mastering them in isolation lets you handle the chains downstream.
Same eight products as in main solution.
Q 6.15
Write the mechanism of the following reaction: n-BuBr + KCNethanol/watern-BuCN.
Concept used.SN2 mechanism for a primary alkyl halide reacting with a strong nucleophile: rate = k [R-X][Nu-]. The nucleophile attacks the C bearing X from the side directly opposite X (backside attack). The three other groups on that C transition from a tetrahedral geometry through a planar trigonal-bipyramidal (TS) and re-pyramidalise on the opposite face (Walden inversion). Single concerted step, no intermediate.
For our system: n-butyl bromide is a 1∘ halide with no β-branching, so the backside is unhindered; CN- is a strong nucleophile (from KCN dissociation in ethanol/water).
[See diagram in the PDF version]
Step 1 (only step). CN- (specifically its carbon end) approaches the C of n-BuBr from the side opposite to Br. A new C-CN bond starts to form while the C-Br bond starts to break, in concert.
Transition state. The C is sp2-like; the three remaining groups (one H from each C-H, and the propyl chain) lie in a plane through the C; CN and Br sit on either side along the reaction axis. Both partial bonds carry partial negative charge.
Products. The Br- ion departs with the bond pair; the C re-pyramidalises on the opposite face. The result is n-butyl cyanide (pentanenitrile) with the C's stereochemistry inverted.
Rate law. rate = k [C4H9Br][CN-]. First order in each, hence ``bimolecular nucleophilic substitution'', SN2.
Why SN2 and not SN1?n-butyl bromide is a primary halide; the primary carbocation CH3CH2CH2CH2+ is highly unstable, so the SN1 pathway does not compete.
SN2 mechanism: single concerted step, backside attack of CN- on the 1∘ C, Walden inversion, rate = k [RBr][CN-].
RG
Rohit Gupta
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Picture-first. Walden's inversion is the visual signature of SN2: think ``umbrella inversion'' at the carbon. The three non-leaving groups pass through a planar arrangement and emerge on the opposite side.
Energy-profile perspective. Plot energy vs. reaction coordinate: starts at the reactant level (R-Br + CN-), rises smoothly to a single transition state (the trigonal-bipyramidal arrangement with partial bonds to both Nu and LG), and falls to the product level (R-CN + Br-). Only one maximum, no intermediate. Contrast with SN1 (two maxima, a carbocation valley between them).
Identify the substrate as primary, the nucleophile as strong (CN-); both push toward SN2.
Draw the curly arrow from the C of CN- to the C bearing Br; simultaneously draw the curly arrow from the C-Br bond out onto Br.
In the transition state, show the three remaining substituents (C3H7, H, H) flattened in a plane through C, with partial bonds to both incoming CN and outgoing Br.
Complete the inversion: the CN group is now on the opposite side from where Br was. Since the C in this substrate is not a stereocentre (two H's on it), the inversion is not observable here, but in a chiral case it would invert configuration.
Confirm by the rate law: rate = k[RBr][CN-] is bimolecular.
Alternative mechanism check. An SN1 pathway would require n-BuBr to ionise to n-Bu+ first. Primary carbocation CH3CH2CH2CH2+ has no π-stabilisation and lies ∼ 280 kJ/mol above the ionised state of a tertiary carbocation. The ionisation step is forbiddingly slow, so SN1 does not compete.
Stereochemistry test for SN2. If the substrate were chiral (e.g. (R)-2-bromobutane), SN2 would invert the configuration to give (S)-2-cyanobutane. This single observation (inversion, not retention, not racemisation) is what distinguishes SN2 from SN1 experimentally.
Why this matters. Backside attack with Walden inversion is one of the most diagnostically clean reactions in organic chemistry. It generalises: any bimolecular nucleophilic substitution at sp3 C inverts the configuration. Modern synthesis uses this for stereocontrolled construction of stereocentres (Mitsunobu reaction, SN2 ring-opening of epoxides with full inversion).
Same: SN2, single step, backside attack, inversion, rate = k[RBr][CN-].
Q 6.16
Arrange the compounds of each set in order of reactivity towards SN2 displacement:
(i) 2-Bromo-2-methylbutane, 1-Bromopentane, 2-Bromopentane;
(ii) 1-Bromo-3-methylbutane, 2-Bromo-2-methylbutane,
(ii) 2-Bromo-3-methylbutane;
(iii) 1-Bromobutane, 1-Bromo-2,2-dimethylpropane,
(iii) 1-Bromo-2-methylbutane, 1-Bromo-3-methylbutane.
Concept used.SN2 reactivity correlates with steric accessibility of the C bearing X. The order is: methyl > 1∘ > 2∘ > 3∘. Among 1∘ halides, branching at the β-carbon further slows the reaction. A neopentyl-type β-branch is the slowest of all primary substrates because it blocks the backside.
Structural observation. Reactivity in SN2 is a steric contest. Rank by (a) class of substrate at α, (b) crowding at β for ties.
Quantitative scale. Relative SN2 rates (typical C-Br, NaOEt in EtOH, set methyl = 1): methyl ≈ 1, ethyl (1∘) ≈ 0.03, 1∘ with β-methyl (isobutyl) ≈ 4 × 10-4, neopentyl (β-tBu) ≈ 4 × 10-6. The drop is dramatic — neopentyl is slower than the corresponding tertiary halide for SN2!
Class hierarchy: methyl > 1∘ > 2∘ > 3∘. Three classes in (i), three in (ii); rank as is.
In (iii), all are 1∘, so move to the β-C. Count methyl substituents on β: 1-Br-butane has 0; 1-Br-3-methylbutane has 0 at β (branch at γ); 1-Br-2-methylbutane has 1 methyl at β; neopentyl has 3 at β. Order is the reverse of β-substitution.
Confirm by mental drawing of the TS for each: more substituents on the β-C means more atoms in the way of the incoming Nu.
Visualisation. Picture the SN2 transition state as a hand reaching from behind the C, while the other three substituents lie flat. For methyl, only three small H's are flat — easy. For neopentyl, three methyls hang off the C next to the planar layer and actively block the approach path of the incoming Nu.
γ-branching is fine. 1-Bromo-3-methylbutane has a branch at γ (two carbons from Br), which is far enough that it does not crowd the backside; its rate is essentially the same as that of n-pentyl bromide. Only β (and α) branching matters.
Concept linkage. The same steric reasoning reverses sign for SN1: tertiary halides ionise fastest because the more substituted carbocation is more stable. Reactivity scales opposite to SN2: 3∘ > 2∘ > 1∘ > methyl.
Why this matters. Steric reasoning is the only tool you need to rank SN2 rates within a homologous series. The same ``count substituents at β'' technique solves any JEE/CBSE ranking question on SN2 within a closed family of primary halides.
Same three orderings as the main solution.
Q 6.17
Out of C6H5CH2Cl and C6H5CHClC6H5, which is more easily hydrolysed by aqueous KOH?
Concept used. Both substrates are benzyl-type chlorides: the C bearing Cl is benzylic. Hydrolysis with aqueous KOH on a benzyl-type halide can proceed by either SN1 or SN2; we expect SN1 character to grow with stabilisation of the resulting benzylic carbocation.
[leftmargin=*,itemsep=1pt]
C6H5CH2Cl: benzyl chloride, 1∘ benzyl. Carbocation PhCH2+ is monobenzylic.
The cation Ph2CH+ is stabilised by resonance into both phenyl rings (extensive +M donation); the cation PhCH2+ is stabilised by resonance into one phenyl. The dibenzylic cation is therefore much more stable.
Since SN1 rate ∝ rate of ionisation ∝ stability of the resulting cation, C6H5CHClC6H5 should hydrolyse faster than C6H5CH2Cl.
!%
[See diagram in the PDF version]
Draw the resonance structures of PhCH2+: positive charge delocalises into the ortho and para positions of the phenyl ring.
Draw the resonance structures of Ph2CH+: positive charge delocalises into both rings, doubling the stabilisation.
Greater delocalisation gives a lower-energy cation, smaller Δ G for ionisation, and a faster SN1 hydrolysis.
Conclude: Ph2CHCl (benzhydryl chloride) is hydrolysed more easily than PhCH2Cl (benzyl chloride).
C6H5CHClC6H5 (benzhydryl chloride) is more easily hydrolysed: the resulting carbocation Ph2CH+ is stabilised by resonance into two phenyl rings, lowering the activation barrier for SN1 ionisation.
IN
Ishita Nair
M.Sc Chemistry, IIT Kanpur
Verified Expert
Strategic angle. The fast-hydrolysis question on benzyl-type halides is always answered by counting cation resonance forms.
Resonance counting recipe. For each candidate carbocation, draw the structure with + on C, then push every adjacent π system or lone pair toward the +. Count distinct contributors that place + on different atoms. For PhCH2+: + on the benzylic C, then + on o (2 of these), then + on p, then back to benzylic — 4 contributors. For Ph2CH+: + on the central C, then on o/p of each of the two phenyl rings — 7 contributors. Roughly twice the delocalisation, much more stable.
In aqueous KOH, polar protic solvent stabilises the ionisation intermediate. Both substrates can in principle undergo SN1.
PhCH2+ has one phenyl ring to delocalise into; the resonance contributors place positive charge at the ortho (2 positions) and para of the ring.
Ph2CH+ has two phenyl rings; positive charge delocalises across both, doubling the stabilising resonance.
Lowering the carbocation energy lowers Δ G for ionisation; rate goes up.
Hence Ph2CHCl reacts faster than PhCH2Cl with aqueous KOH.
Mechanism perspective. Hydrolysis proceeds via SN1: rate-determining ionisation gives the carbocation, which is then trapped by H2O (or OH-) in a fast step. So the rate depends only on [RCl], not on [OH-]. This means the rate ratio Ph2CHCl/PhCH2Cl would be the same in pure water or in dilute KOH.
Triphenyl extension. For triphenylmethyl (trityl) chloride Ph3CCl, the cation is stabilised by three rings — exceptionally stable. Trityl chloride hydrolyses in pure water at room temperature, whereas PhCH2Cl needs warm aqueous KOH.
Concept linkage. The same logic explains why allyl, benzyl, and especially diphenyl/triphenyl methyl substrates react via SN1 even when they are primary or secondary — substantial cation π-stabilisation overrides the standard 1∘ < 2∘ < 3∘ progression based purely on inductive effects.
Why this matters. The trick is to spot that we are comparing carbocation stabilities, not steric accessibilities; benzhydryl wins decisively because of double resonance. CBSE short-answer questions on ``which halide hydrolyses faster?'' are almost always carbocation stability problems in disguise.
p-Dichlorobenzene has higher m.p. than those of o- and m-isomers. Discuss.
Concept used. For molecules of comparable polarity and molar mass, the melting point is governed primarily by crystal packing: how efficiently the molecules stack in the solid state. High symmetry allows molecules to pack tightly into the unit cell, increasing intermolecular contact and lattice energy, and raising the melting point. The boiling point, by contrast, depends mostly on the dipole moment and van der Waals attractions in the liquid.
Symmetry argument.p-Dichlorobenzene has a C2h symmetry: the molecule is centrosymmetric. The two C-Cl dipoles are antiparallel and cancel (μ = 0 D). Molecules are flat, linear-shaped and pack into a highly ordered, dense crystal.
The o- and m-isomers are not centrosymmetric and have non-zero μ (o- μ ≈ 2.5 D, m- μ ≈ 1.7 D). Their less symmetric shapes pack less efficiently in the solid state.
Numerical comparison.
[leftmargin=*]
o-dichlorobenzene: m.p. ≈ -17∘C, b.p. ≈ 180∘C.
m-dichlorobenzene: m.p. ≈ -25∘C, b.p. ≈ 173∘C.
p-dichlorobenzene: m.p. ≈ 53∘C, b.p. ≈ 174∘C.
The p-isomer's m.p. is dramatically higher.
Why b.p. doesn't follow the m.p. trend.o-Dichlorobenzene has the highest dipole moment and therefore the highest b.p. p-Dichlorobenzene's μ = 0 D, so its b.p. is among the lowest. The discrepancy between b.p. and m.p. trends is precisely the ``packing vs. dipole'' distinction.
[See diagram in the PDF version]
p-dichlorobenzene's high symmetry and zero dipole let its molecules pack into a tighter, more ordered crystal lattice, giving the highest melting point of the three isomers.
MS
Meera Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Structural observation. M.p. is a crystal property; b.p. is a liquid–gas property. They can move in opposite directions when symmetry and dipole disagree.
Sketch of the crystal-packing argument. In a molecular crystal, the lattice energy comes from (a) dispersion (London) forces between heavy atoms, (b) dipole–dipole alignment, and (c) shape complementarity (how snugly the molecules fit). Higher shape symmetry = more efficient packing density = more neighbouring contacts per molecule = larger total lattice attraction. Even though p-dichlorobenzene has zero dipole, its shape (linear-rod) packs into a denser unit cell than the bent shapes of o- and m-isomers.
Sketch each isomer; mark the two C-Cl dipoles and the molecular symmetry axis.
Only the para-isomer is centrosymmetric; its μ = 0 and it has the highest symmetry.
In the solid, high symmetry means molecules stack like flat coins into close-packed columns. Lattice energy is therefore largest for the para-isomer.
Heating breaks the lattice; the para lattice requires the most heat to disrupt, hence the highest m.p.
The o- and m-isomers have higher dipole moments but poorer packing; net result is a lower m.p. but a competitive (sometimes higher) b.p.
Boiling-point cross-check.o-dichlorobenzene: b.p. 180∘C (μ ≈ 2.5 D, highest); m-: b.p. 173∘C (μ ≈ 1.7 D); p-: b.p. 174∘C (μ = 0 D). B.p. correlates with dipole moment, not with crystal packing — exactly the opposite trend to m.p.
Concept linkage. The same idea generalises to n-alkanes (even-numbered alkanes pack better than adjacent odd ones, so their m.p. is locally higher), to symmetric vs. asymmetric isomers of disubstituted aromatics in general, and to fullerenes (C60 packs like a sphere, very high m.p.).
Exam-tip. If asked ``why is the m.p. of A higher than that of B?'', invoke crystal-packing/symmetry. If asked ``why is the b.p. of A higher than that of B?'', invoke dipole moment and/or molecular mass (dispersion).
Why this matters. A classic two-mark NCERT question. Use symmetry + packing for m.p.; dipole + vdW for b.p. The counter-intuitive result that the most symmetric (zero-dipole) isomer has the highest m.p. trains the eye to separate ``melting'' (crystal → liquid) from ``boiling'' (liquid → gas) in problems on physical properties.
Higher symmetry → tighter crystal → higher m.p. of p-dichlorobenzene compared with the o- and m-isomers.
Q 6.19
How can the following conversions be carried out?
(i) Propene to propan-1-ol; (ii) Ethanol to but-1-yne; (iii) 1-Bromopropane to 2-bromopropane; (iv) Toluene to benzyl alcohol; (v) Benzene to 4-bromonitrobenzene; (vi) Benzyl alcohol to 2-phenylethanoic acid; (vii) Ethanol to propanenitrile; (viii) Aniline to chlorobenzene; (ix) 2-Chlorobutane to 3,4-dimethylhexane; (x) 2-Methyl-1-propene to 2-chloro-2-methylpropane; (xi) Ethyl chloride to propanoic acid; (xii) But-1-ene to n-butyliodide; (xiii) 2-Chloropropane to 1-propanol; (xiv) Isopropyl alcohol to iodoform; (xv) Chlorobenzene to p-nitrophenol; (xvi) 2-Bromopropane to 1-bromopropane; (xvii) Chloroethane to butane; (xviii) Benzene to diphenyl; (xix) tert-Butyl bromide to isobutyl bromide; (xx) Aniline to phenylisocyanide.
Concept used. Twenty conversions, each a chain of named reactions: hydroboration–oxidation, Wurtz, Sandmeyer, hydrolysis, Williamson, Kolbe nitrile, carbylamine, etc.
Halide-shift pattern explained. Conversions (iii), (xvi), (xix) all involve relocating a halogen from one carbon to a neighbouring carbon. The universal recipe is: (1) eliminate to the alkene with alc. KOH, (2) re-add HX. If Markovnikov gives the desired product, use plain HX; if anti-Markovnikov is needed, use HX/peroxide. This two-step ``halide-shift'' is the only way to relocate a halogen in NCERT-level chemistry.
Walk through each in ∼ 1 minute: identify the functional group change, pick the standard reagent.
Pay attention to directing effects in aromatic substitutions (Cl, Br are o,p-directors; NO2 is a m-director).
For halide-shift problems (xiii, xvi, xix), the recipe is always: E2 to give alkene, then add HX with peroxide if you want anti-Markovnikov.
Order of operations in (v) and (xv). For C6H6 → 4-bromonitrobenzene, brominate before nitrating because Br is o,p-directing but NO2 is m-directing — the order determines which isomer dominates. For chlorobenzene → 4-nitrophenol, nitrate first to activate the ring toward nucleophilic substitution of Cl; without the para NO2 the Cl would not be hydrolysed under reasonable conditions.
Wurtz vs. Wurtz–Fittig in (ix), (xvii), (xviii). Wurtz: R-X + R-X + 2 Na -> R-R + 2 NaX (alkyl coupling). Wurtz–Fittig: Ar-X + R-X + 2 Na -> Ar-R + 2 NaX (mixed alkyl-aryl) or 2 Ar-X + 2 Na -> Ar-Ar + 2 NaX (aryl-aryl). For (xviii) we use the latter on PhBr to give biphenyl.
Spotting Sandmeyer in (viii). Aniline (ArNH2) → aryl halide requires the diazonium intermediate Ar-N2+X-, which is then displaced by CuX (Sandmeyer) or by aqueous HCl boil (Gattermann). Aniline can never be converted directly to chlorobenzene with HCl or NaCl.
Why this matters. Master the table of ``reaction class → reagent'' and you can produce these from a clean slate. NCERT Q 6.19 with its twenty parts is the chapter's flagship pattern-drill; working through it builds the foundation for organic-chemistry retrosynthesis throughout class 12. JEE-Advanced 1-mark MCQs are nearly always drawn from this list of named transformations.
Same twenty reagent sequences.
Q 6.20
The treatment of alkyl chlorides with aqueous KOH leads to the formation of alcohols but in the presence of alcoholic KOH, alkenes are major products. Explain.
Concept used. Aqueous KOH and alcoholic KOH differ in two key ways:
[leftmargin=*,itemsep=1pt]
Active species. In aqueous KOH, the dominant species is OH- (a strong nucleophile, small/hard). In alcoholic KOH (KOH dissolved in ethanol), the active species is C2H5O^- (ethoxide), which is a bulkier and stronger base than OH-.
Solvent. Water is a polar protic solvent that stabilises OH- and the charge build-up in the substitution transition state; ethanol is also protic but less so, and it solvates ethoxide less tightly.
The two channels are: aligned Substitution ($SN2$ or $SN1$): &R-Cl + OH- -> R-OH + Cl-,
Elimination ($E2$ or $E1$): &R-CHR′-CH2-Cl + OEt-
& -> R-CH=CH-R′ + EtOH + Cl-. aligned
Substitution wins when the base is a good nucleophile (small, unhindered) and the substrate is unhindered; elimination wins when the base is bulky/stronger and the substrate has accessible β-H's.
Aqueous KOH. The reactive species is OH-, a small, strong nucleophile and a weaker base than ethoxide. Attack on the C-Cl carbon is fast, giving an alcohol via SN pathway. Water-mediated stabilisation of the polar transition state pushes the system toward substitution.
Alcoholic KOH. The reactive species is C2H5O- (ethoxide), bulkier and stronger as a base than hydroxide. It preferentially abstracts a β-H from R-Cl in a concerted E2 step: H-C-C-Cl EtO-C=C + EtO-H + Cl-. Because EtO- is bulky, it is less efficient at the crowded backside attack on C, so it abstracts the more accessible β-H instead.
Saytzeff vs Hofmann. EtO- is not bulky enough to enforce Hofmann; the elimination still gives the Saytzeff (most substituted) alkene as the major product.
Summary.
[leftmargin=*]
Aqueous KOH → alcohol (substitution dominates).
Alcoholic KOH → alkene (elimination dominates).
Aqueous KOH provides OH-, a strong but small nucleophile that prefers substitution to give R-OH. Alcoholic KOH provides ethoxide (C2H5O-), a bulkier and stronger base that prefers elimination to give the alkene.
SC
Siddharth Chatterjee
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Strategic angle. Same reagent, two different solvents, two different products. The discriminator is which species is solvated vs. free.
Acid–base picture of solvent effect. In water, the equilibrium KOH(aq) <=> K+ + OH- favours the right completely; OH- is the only meaningful nucleophile. In ethanol, KOH dissolves and also establishes OH- + C2H5OH <=> C2H5O- + H2O, which has Keq ≈ 1 because pKa(H2O)≈ 15.7 and pKa(EtOH)≈ 16. So a mixture of OH- and C2H5O- is present in alcoholic KOH. Ethoxide, being slightly bulkier and slightly stronger, dominates the elimination channel.
In water, OH- is the active species: small, strong nucleophile, moderate base. With a primary or secondary alkyl chloride, it does SN to give the alcohol.
In ethanol, KOH dissolves with deprotonation of ethanol and C2H5O- (ethoxide) becomes the active species. Ethoxide is bulkier and a stronger base, so it abstracts a β-H (E2) in preference to attacking the C.
Polar protic solvents (water more so than ethanol) stabilise the build-up of charge in the substitution TS; less polar protic solvents (ethanol) tilt the balance toward elimination.
Temperature also matters. Heating (Δ) favours elimination over substitution because E2 has a higher activation entropy (more degrees of freedom in the TS) and is favoured at higher T. So warm alcoholic KOH gives the cleanest alkene yield.
Substrate sensitivity. The substitution-vs- elimination balance also depends on substrate class: 1∘ halides prefer SN2 even with bulky bases; 3∘ halides prefer E2 even with small bases; 2∘ halides are in the borderline zone where solvent choice dictates the outcome.
Why this matters. The same idea generalises: choose aqueous for substitution, alcoholic for elimination, bulky alkoxide (t-BuOK) for Hofmann elimination. The trio ``aqueous KOH, alcoholic KOH, t-BuOK'' covers nearly every substitution–elimination scenario in class 12 organic chemistry.
Primary alkyl halide C4H9Br (a) reacted with alcoholic KOH to give compound (b). Compound (b) is reacted with HBr to give (c) which is an isomer of (a). When (a) is reacted with sodium metal it gives compound (d), C8H18, which is different from the compound formed when n-butyl bromide is reacted with sodium. Give the structural formula of (a) and write the equations for all the reactions.
Concept used.
[leftmargin=*,itemsep=1pt]
Primary C4H9Br candidates: 1-bromobutane (n-BuBr) and 1-bromo-2-methylpropane (isobutyl bromide).
Wurtz on n-BuBr gives n-octane. The problem says (d) is different from what n-BuBr gives, so (a) cannot be n-BuBr. Hence (a) = isobutyl bromide.
Alcoholic KOH on isobutyl bromide eliminates to give 2-methylpropene (isobutylene).
HBr Markovnikov on 2-methylpropene gives (CH3)3CBr, the 3∘ tert-butyl bromide. This is a constitutional isomer of (a).
Wurtz on isobutyl bromide gives 2,5-dimethylhexane, a C8H18 compound different from n-octane.
Identify (a). (a) is primary, and Wurtz on (a) gives a C8H18 hydrocarbon different from n-octane. So (a) =isobutyl bromide, (CH3)2CHCH2Br.
Strategic angle. The two clues that nail down (a) are: (1) primary halide, (2) Wurtz product differs from n-octane. Combined, they force (a) = isobutyl bromide.
Logic-puzzle framework. This is a classic ``identify the compound from the reactions'' puzzle. Whenever an NCERT problem gives you a series of clues, write the candidates that satisfy each clue, then take the intersection. Here, ``primary C4H9Br'' has 2 candidates; ``Wurtz ≠n-octane'' has 1 candidate (since n-BuBr Wurtz =n-octane). The intersection is unique.
Filter 1: primary C4H9Br: candidates are n-BuBr and isobutyl bromide.
Filter 2: Wurtz on (a) gives a C8H18 different from the Wurtz product of n-BuBr (=n-octane). So (a) is isobutyl bromide.
Alc. KOH gives the only Saytzeff alkene available: 2-methylprop-1-ene.
HBr Markovnikov: H to the less substituted C (=CH2), Br to the more substituted C (C(CH3)2). Product: t-BuBr, an isomer of (a). Tick.
Wurtz coupling: two molecules of (CH3)2CHCH2Br couple at the CH2Br centres to give (CH3)2CHCH2-CH2CH(CH3)2 = 2,5-dimethylhexane.
Counter-check (a) by self-consistency. If (a) were n-BuBr instead, the Wurtz product would be n-octane (CH3(CH2)6CH3) — but the problem explicitly says (d) is different from that. So n-BuBr is excluded. The alc. KOH product of n-BuBr is but-1-ene, whose Markovnikov HBr addition gives 2-bromobutane (not an isomer of (CH3)2CHCH2Br in the sense the question intends; it is an isomer of n-BuBr though). Confusing, which is why the Wurtz check is the cleaner filter.
Concept linkage. This problem packs four named reactions into one identification puzzle: dehydrohalogenation (E2 Saytzeff), Markovnikov HX addition, Wurtz coupling. Each clue is itself a one-mark sub-question. CBSE 5-mark questions of this format are routine.
Why this matters. A logic puzzle: each clue removes one candidate. The skill of intersecting structural clues (primary, halide, Wurtz product different from a specific isomer) is exactly the analytical thinking that JEE and AIIMS test repeatedly. The same puzzle template applies to any ``mystery compound A reacts as follows; identify A'' question.
Same (a)–(d) assignments as in main solution.
Q 6.22
What happens when
(i) n-butyl chloride is treated with alcoholic KOH; (ii) bromobenzene is treated with Mg in the presence of dry ether; (iii) chlorobenzene is subjected to hydrolysis; (iv) ethyl chloride is treated with aqueous KOH; (v) methyl bromide is treated with sodium in the presence of dry ether; (vi) methyl chloride is treated with KCN?
Quick reading. Six small reactions, one named transformation each. Identify the reagent class, predict the product.
Reagent → named-reaction table.Alc. KOH→E2 (dehydrohalogenation); Mg/dry ether→ Grignard; NaOH at 623 K/300 atm→ Dow process (phenol from chlorobenzene); Aq. KOH→SN (alcohol); Na/dry ether→ Wurtz coupling; KCN→ ambident, C-attack, nitrile.
(i) Alc. KOH =E2⇒ alkene. The only Saytzeff product from n-BuCl is but-1-ene.
(ii) Mg in dry ether = Grignard formation; C6H5MgBr is one of the most useful carbanion equivalents.
(iii) Chlorobenzene is essentially inert to ordinary NaOH because the C-Cl bond has partial double-bond character (resonance) and the ring C is sp2. Industrial Dow process (623 K, 300 atm NaOH) gives phenol.
(iv) Aq. KOH =SN on a primary halide → alcohol. Ethyl chloride gives ethanol.
(v) Na in dry ether = Wurtz. Methyl bromide gives CH3CH3 (ethane).
Why chlorobenzene resists hydrolysis (iii). Three factors: (a) sp2 C is more electronegative than sp3, so the C-Cl bond is shorter and stronger; (b) lone-pair donation from Cl into the ring gives partial double-bond character to C-Cl; (c) the backside of the sp2 C is occupied by the π-cloud, so SN2 backside attack is impossible. The only way around all three is the harsh Dow conditions, which proceed via a benzyne intermediate.
Grignard's silent partner is dry ether. The ether oxygen lone pairs solvate Mg^2+, stabilising the reagent in solution. THF is even better than diethyl ether for unreactive aryl halides; without coordinating solvent, the Grignard would not form.
Concept linkage. (i) connects to Q 6.20; (ii) to Q 6.12; (iii) to the haloarenes section discussion; (iv) also to Q 6.20; (v) to Q 6.21's Wurtz argument; (vi) to Q 6.8's ambident nucleophile analysis. The six parts of Q 6.22 effectively summarise the entire chapter in capsule form.
Why this matters. Six standard reactions in six lines. Master the reagent → outcome mapping. In CBSE board exams, ``what happens when X is treated with Y?'' is the most frequent 2-mark question format; six such pairs in one question is essentially a comprehensive end-of-chapter test.
Same six products as in main solution.
More Haloalkanes and Haloarenes Chemistry Class 12 Resources
Haloalkanes and Haloarenes Class 12 Chemistry NCERT Solutions FAQs
Ques. Where can I download Haloalkanes and Haloarenes Class 12 Chemistry NCERT Solutions PDF?
Ans. You can download the Haloalkanes and Haloarenes Class 12 Chemistry NCERT Solutions PDF directly from this page. Both Normal and HD versions are available, and both are free.
Ques. Are the Haloalkanes and Haloarenes NCERT Solutions aligned with the 2026-27 NCERT?
Ans. Yes. This page reflects the current 2026-27 syllabus for Class 12 Chemistry. The new edition keeps the core sub-topics (preparation, SN1/SN2, E1/E2, haloarene electrophilic substitution) intact and only the bulk industrial applications of polyhalogen compounds are trimmed.
Ques. How many pages is the Class 12th Chemistry Haloalkanes and Haloarenes NCERT Solutions PDF?
Ans. The NCERT Solutions PDF runs approximately 34 pages and covers all 32 questions (10 in-text + 22 back exercise), each with the mechanism, intermediate, and product shown on separate lines.
Ques. What is the difference between SN1 and SN2 mechanisms in Haloalkanes?
Ans. SN1 is a two-step unimolecular mechanism via a carbocation intermediate, favoured by tertiary substrates and polar protic solvents, and gives racemised product. SN2 is a single-step bimolecular mechanism with a backside attack, favoured by primary substrates and polar aprotic solvents, and gives inversion of configuration. CBSE awards two marks for stating the order, kinetics, and stereochemistry side by side.
Ques. Why are haloarenes less reactive than haloalkanes toward nucleophilic substitution?
Ans. The C-X bond in haloarenes has partial double-bond character due to resonance with the benzene ring, making it shorter and stronger than in haloalkanes. The sp2 carbon is also more electronegative, holding the halogen more tightly. Both factors make nucleophilic substitution difficult without forcing conditions.
Ques. How many questions does the Haloalkanes and Haloarenes NCERT chapter contain?
Ans. The chapter has 10 in-text questions and 22 back-exercise questions, for a total of 32 questions. All 32 are solved in the downloadable PDF on this page.
Ques. How much time should I spend on Haloalkanes and Haloarenes for the Class 12 Boards?
Ans. Plan for about 9 to 11 hours of focused practice spread across four sessions, covering nomenclature, preparation, mechanisms, and conversions. SN1 vs SN2 reasoning deserves the most time as it appears in nearly every board paper.
Ques. Is Haloalkanes and Haloarenes important for JEE Main and NEET?
Ans. Yes. Haloalkanes and Haloarenes carries about 3 to 4 percent weightage in JEE Main and 2 to 3 questions per year in NEET. SN1/SN2 reactivity order, optical activity of chiral haloalkanes, and named reactions (Sandmeyer, Wurtz-Fittig, Finkelstein) are the most frequently tested sub-topics across both exams.
Ques. What is the Saytzeff rule and how does it differ from the Hofmann rule?
Ans. Saytzeff's rule states that in β-elimination with a small base (alc. KOH, ethoxide), the major alkene is the more substituted (more stable) one. Hofmann's rule applies when a bulky base (potassium tert-butoxide) is used; the less-substituted alkene becomes the major product because the bulky base prefers the less hindered β-H. Confusing Saytzeff vs Hofmann is a 2-mark slip in CBSE.
Ques. What is the Kharasch effect (peroxide rule) for the addition of HBr to alkenes?
Ans. In the presence of peroxides (R-O-O-R), HBr adds to an unsymmetrical alkene in an anti-Markovnikov manner: the Br goes to the carbon bearing more hydrogens. This Kharasch peroxide effect operates only with HBr (not HCl or HI) because only the Br radical chain is energetically favourable. Without peroxides, the regular Markovnikov product dominates.
Ques. How do you assign R and S configuration to a chiral haloalkane?
Ans. Rank the four groups on the chiral carbon by CIP priority (higher atomic number wins; for ties, look at next-shell atoms). Orient the molecule so the lowest-priority group points away. If the remaining three groups go 1 → 2 → 3 clockwise, the centre is R (rectus); anti-clockwise is S (sinister). For 2-bromobutane, Br > CH2CH3 > CH3 > H gives the assignment.
Ques. What is the role of anhydrous ZnCl2 in the Lucas test for alcohols?
Ans. Anhydrous ZnCl2 acts as a Lewis-acid catalyst in the Lucas reagent (conc. HCl + anhydrous ZnCl2) used to distinguish 1°, 2°, and 3° alcohols. The ZnCl2 protonates and weakens the C-O bond, helping it ionise. 3° alcohols react immediately (cloudy at once), 2° alcohols take 5-10 minutes, and 1° alcohols do not react at room temperature. The product is the alkyl chloride.
Ques. How are DDT and freons (CFCs) prepared and why are they environmentally hazardous?
Ans. DDT (p,p′-dichlorodiphenyltrichloroethane) is prepared by condensing chlorobenzene with chloral (CCl3CHO) in conc. H2SO4. Freons (CFCs like CCl2F2) are made from CCl4 by Swarts reaction with SbF3/HF. Both bio-accumulate or deplete the ozone layer: DDT is fat-soluble and persists in food chains; CFC radicals catalyse Cl-mediated O3 destruction in the stratosphere. CFCs are banned under the Montreal Protocol; DDT is banned in most countries.
Ques. What is racemization and why does an SN1 reaction give a racemic mixture?
Ans. Racemization is the conversion of an optically active compound into an equal (1:1) mixture of its enantiomers, giving zero net optical rotation (the racemic mixture). In SN1 the C-X bond ionises first, producing a planar sp2 carbocation. The nucleophile can attack from either face of this planar intermediate with equal probability, yielding both (R) and (S) products in equal amounts. The result is a racemic (±) product.
































































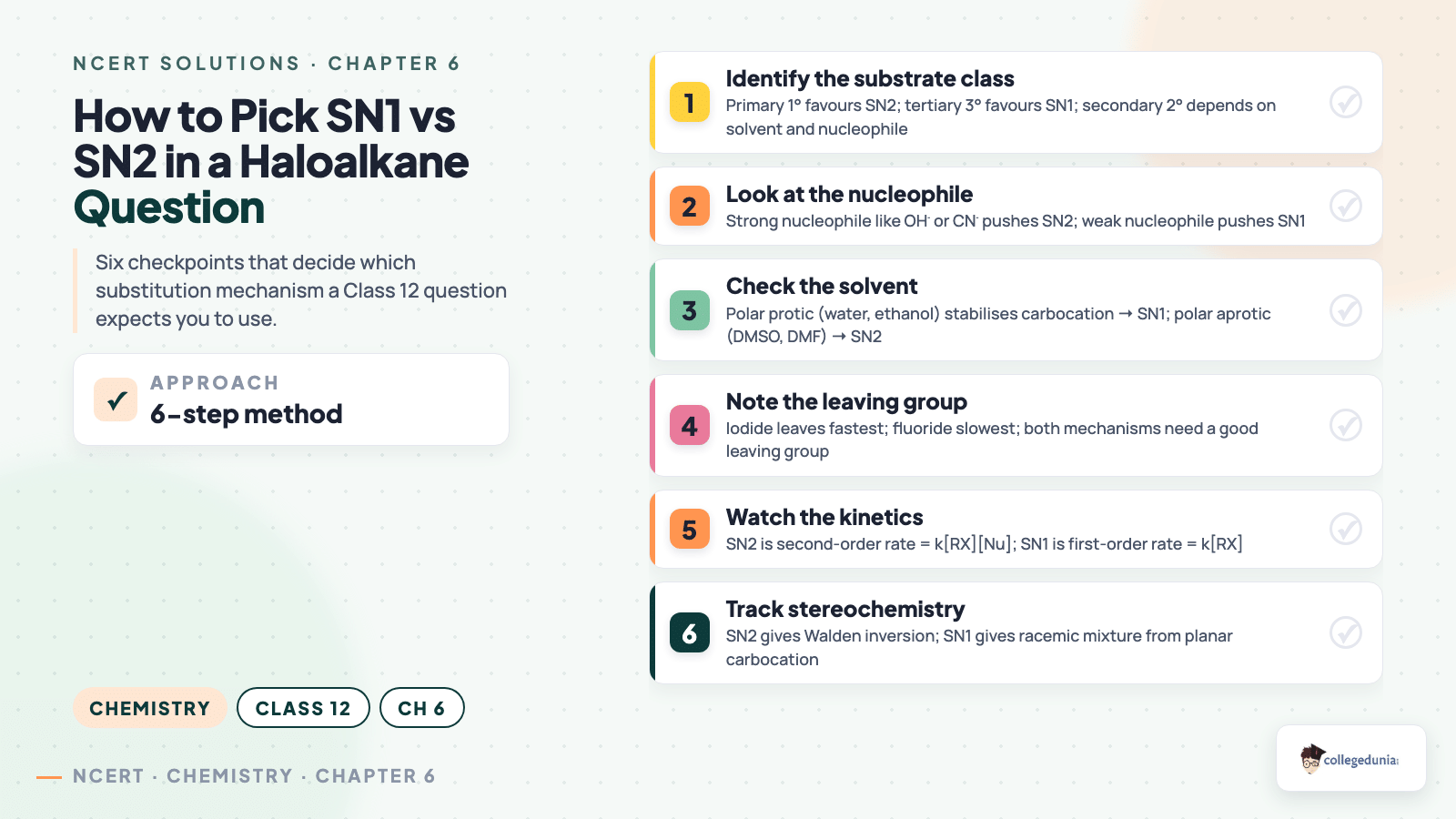



Comments